Team:
Welcome to Phenotype Phebruary Day 4! We’ve discuss phenotyping metabolic diseases (T2DM and T1DM) and a cardiovascular disease (AFib), so let’s turn our attention to a different disease area that is a particularly active focus for many community collaborators - oncology. OHDSI’s Oncology workgroup has made good progress in thinking about advances in the OMOP CDM and OHDSI vocabularies to accommodate the study of cancers and their treatments. Another key opportunity, that I know @agolozar is quite keen to lead within the Oncology WG, is developing phenotype algorithms for each cancer target, and evaluating those algorithms across a diverse array of databases that could potentially be used to generate evidence, including administrative claims, electronic health records, specialty oncology EHRs, and cancer registries. Phenotype Phebruary seems the perfect time to get community collaboration toward this objective, starting today with Multiple Myeloma.
Clinical description:
Multiple myeloma is a type of blood cancer that affects plasma cells. Malignant white blood cells develop in bone marrow, suppressing healthy plasma cells that produce antibodies against infection. Malignant plasma cells produce M protein, which can cause tumors, kidney damage, bone destruction and impaired immune function. They also cause decreased production of red blood cells and platelets, which can result in anemia and bleeding.
Multiple myeloma is diagnosed based on plasmacytoma identified on biopsy, >30% malignant plasma cells in bone marrow aspiration, evaluated levels of M protein from protein electrophoresis in the blood or urine, osteolytic lesions observed on imaging, and IgG or IgA antibody levels in the blood. Additional diagnostics tests may include measurement of Beta2-microglobulin level. Management of multiple myeloma typically requires pharmacologic treatment with proteasome inhibitors (including bortezomib, carfilzomib, ixazomib), immunomodulatory drugs (like lenalidomide, pomalidomide, thalidomide), steroids (dexamethasone, prednisone), monoclonal antibodies (such as elotuzumab, daratumumab, isatuximab, belantamab), and chemotherapy (doxorubicin, melphalan, cyclophoshamide, bendamsutine, vincristine). Autologous stem cell transplant may be considered for those eligible. Patients may also be treated with bisphosphonates to reduce risk of bone loss.
Multiple myeloma is more common in men than women, more common in Black or African American than whites, more common in older ages (with most cases occurring after 40 years old)
Phenotype development:
There have been several prior publications studying multiple myeloma in observational data, including a few with validation efforts of phenotype algorithms.
Brandenburg et al, " Validating an algorithm for multiple myeloma based on administrative data using a SEER tumor registry and medical record review", published in Pharmacoepidemiology and Drug Safety in 2019, provides a useful assessment of four alternative phenotype algorithms:

The authors developed and evaluated these four algorithms initially within Henry Ford Health System, then applied the algorithm within the Optum claims database to estimate positive predictive value through source record adjudication of a sample of the cases identified by their preferred algorithm (#2).
I’ll note: @jswerdel included this study in his phenotype algorithm performance benchmark, which he presented at OHDSI2020. His past work on this topic made my live a million times easier as I was putting this together, so big shout out and thanks to Joel for that. I’ll let him comment on the empirical results of applying PheValuator to these definitions.
Here’s how we can implement each of the Branderburg algorithms using ATLAS:
Algorithm 1: “≥2 ICD‐9‐CM codes for multiple myeloma (203.0x) at least 30 days apart” (ATLAS-phenotype link here)
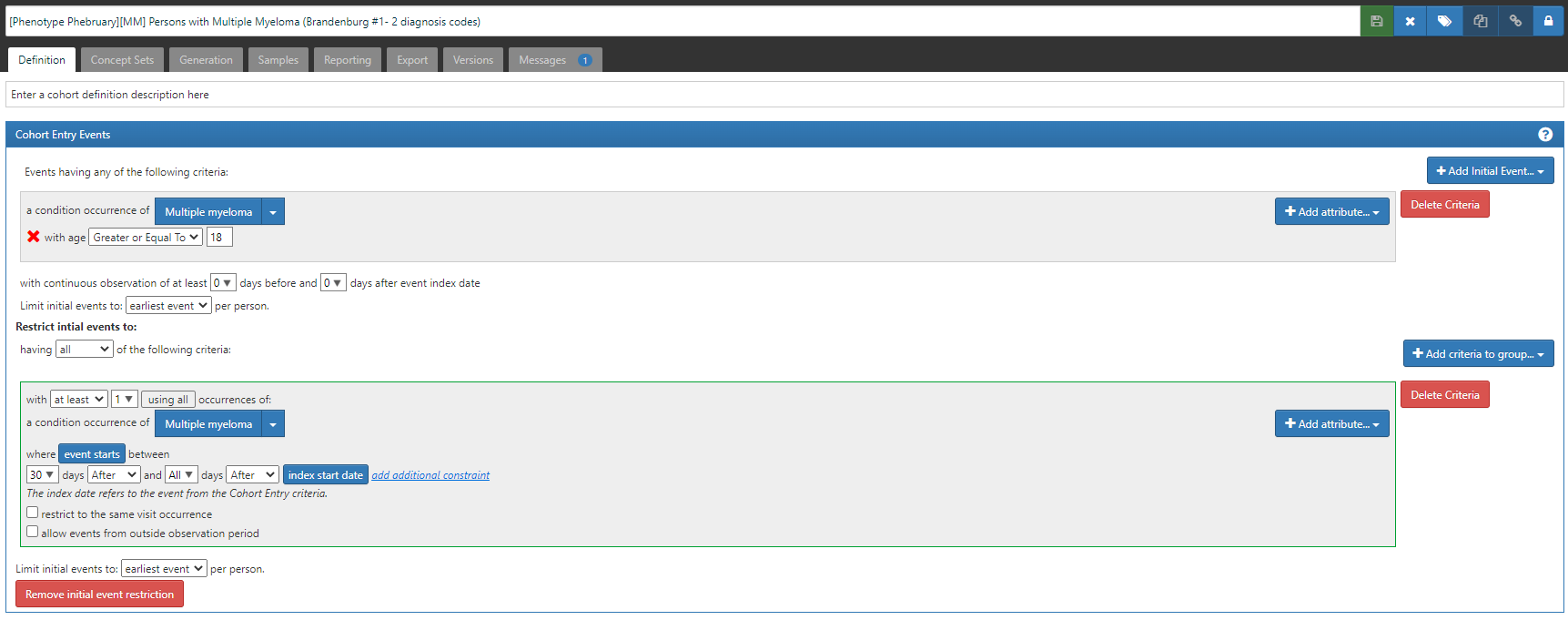
Algorithm 2: “Two‐part algorithm. Both parts of the algorithm are required. Parts 1 and 2 have to be fulfilled separately and sequentially. Part 1: ≥2 ICD‐9‐CM codes for multiple myeloma (203.0x) AND ≥1 procedure code for bone marrow aspirate, biopsy, or interpretation OR ≥1 procedure code for two diagnostic tests (the tests must be different) Part 2: ≥2 ICD‐9‐CM codes for multiple myeloma (203.0x) 5 to 90 days after procedures identified in part 1” (ATLAS-phenotype link here)
Start with algorithm #1, then add this extra clause:
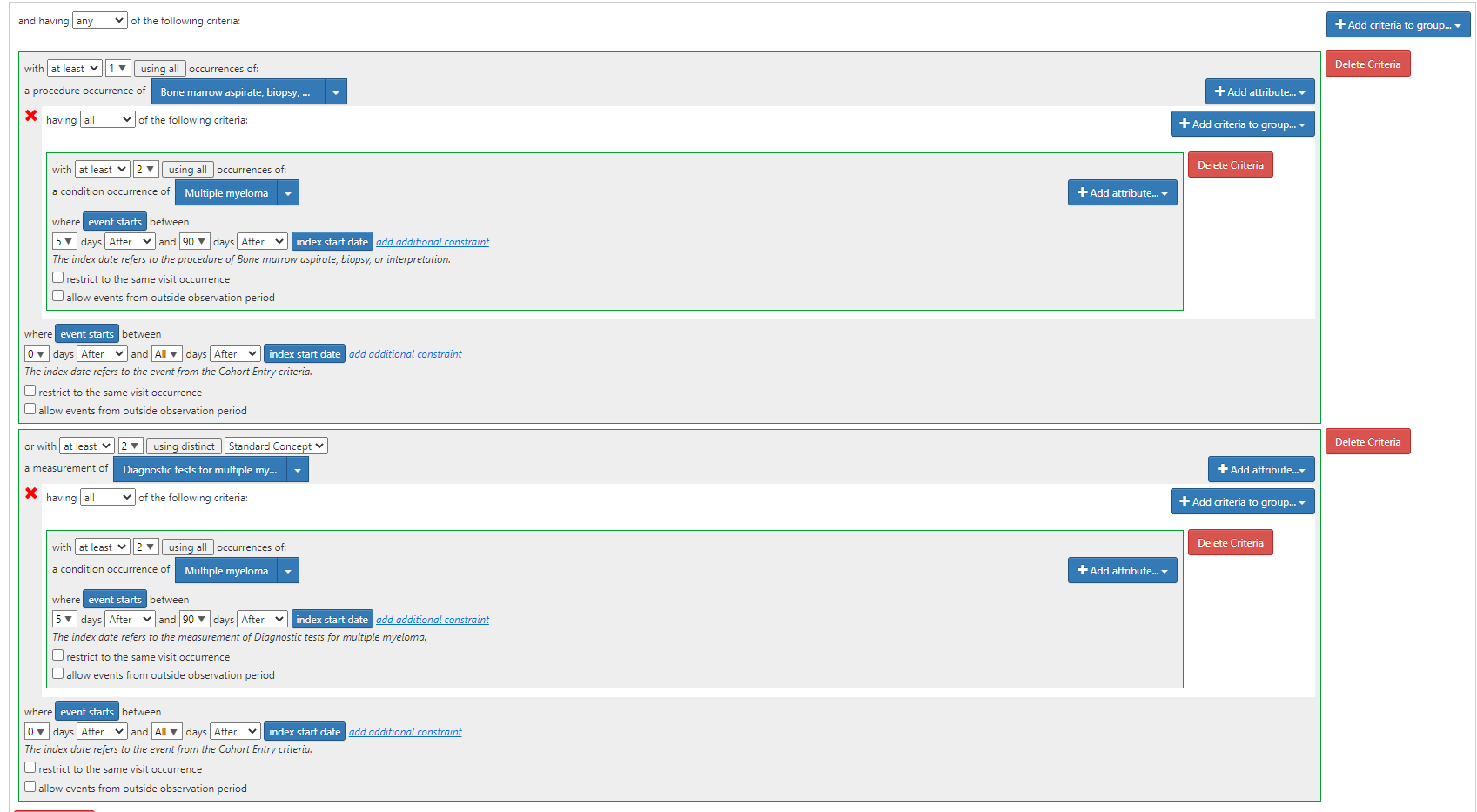
Algorithm 3: “Two‐part algorithm. Both parts of the algorithm are required. Parts 1 and 2 have to be fulfilled separately and sequentially.Part 1: ≥2 ICD‐9‐CM codes for multiple myeloma (203.0x) AND ≥1 procedure code for bone marrow aspirate, biopsy, or interpretation OR ≥1 procedure code for two diagnostic tests (the tests must be different) Part 2: ≥2 ICD‐9‐CM codes for multiple myeloma (203.0x) 5 to 90 days after procedures identified in part 1 AND ≥1 prescription claim for a multiple myeloma therapy” (ATLAS-phenotype link here)
Algorithm 3 amends the clause shown for Algorithm 2 by adding the additional drug requirement:

(note, the drug list used in the publication is likely incomplete, as other drugs are currently in use, including daratumumab, carfilzomib, ixazomib, pomalidomide, elotuzumab, isatuximab, belantamab.
Algorithm 4: “Three‐part algorithm. All parts of the algorithm are required. Parts 1, 2, and 3 have to be fulfilled separately and sequentially.Part 1: ≥2 ICD‐9‐CM codes for multiple myeloma (203.0x) AND ≥1 procedure code for bone marrow aspirate, biopsy, or interpretation OR ≥1 procedure code for two diagnostic tests (the tests must be different) Part 2: ≥2 ICD‐9‐CM codes for multiple myeloma (203.0x) 5 to 90 days after procedures identified in part 1 Part 3: If ≥2 ICD‐9‐CM codes for monoclonal gammopathy of undetermined significance (273.1) are present after parts 1 and 2 of the algorithm are fulfilled, then exclude the patient” (ATLAS-phenotype link here)
I think it’s a nice testament that these fairly complex algorithms, each with their own custom source code lists, could all be modeled directly in ATLAS without any issue. And added bonus, now that they are standardized to OMOP CDM and standard concepts, they’ll work not only for ICD9 codes provided, but can also be extended to other source codes (like ICD10CM to focus on the current myeloma cases in our US databases).
Phenotype evaluation:
Brandenburg et al. used source record verification as their approach to validation in the Optum database, whereby 104 charts were extracted to confirm the diagnosis. Their estimate of PPV was 86%.
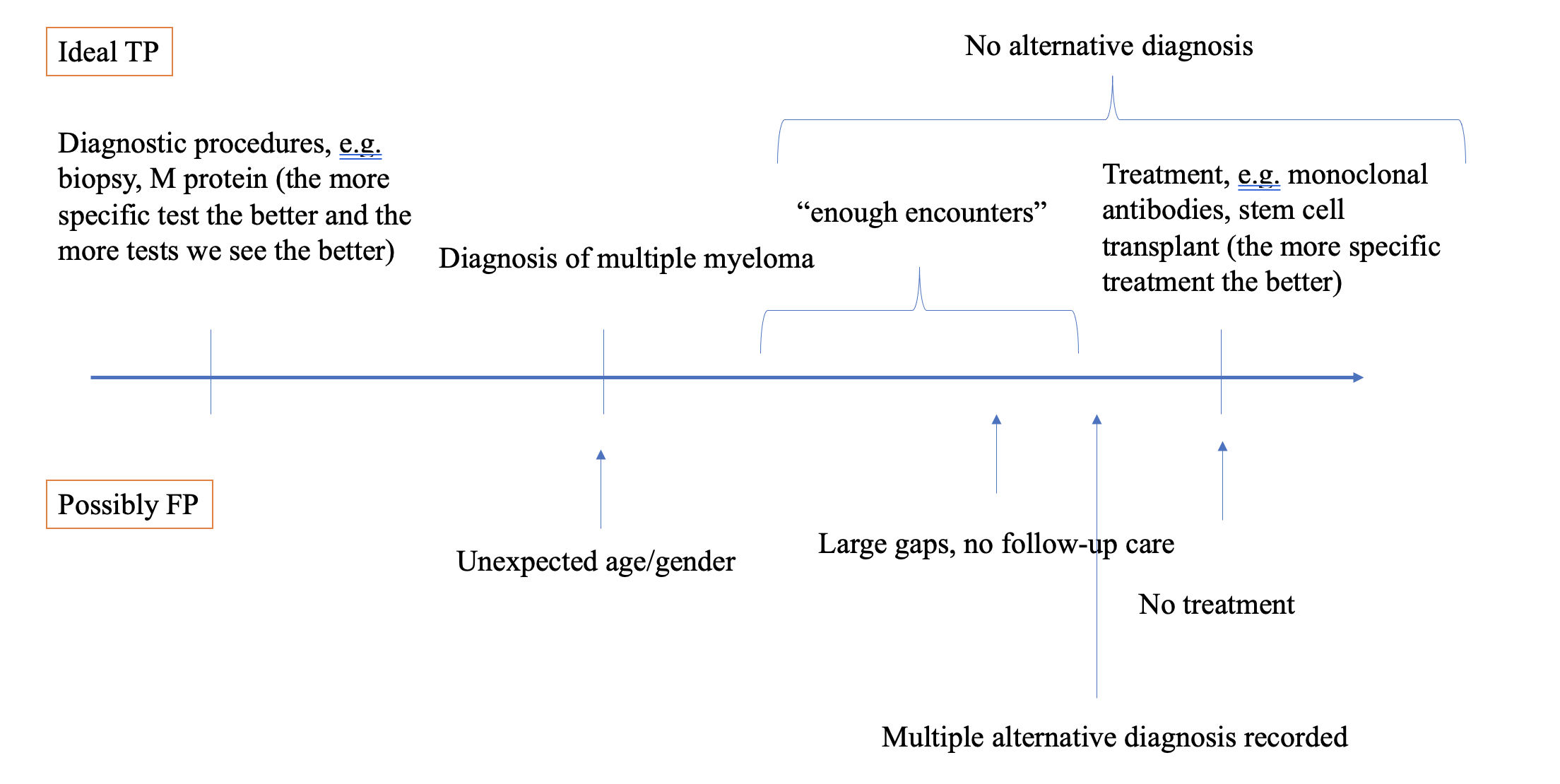
In many databases, source records are not available. In other circumstances, the time and resources associated with manual chart review is prohibitive. We’ve talked about PheValuator and CohortDiagnostics as OHDSI tools that can be used to support the evaluation process. Today, I’d like to highlight how patient profile reviews can be conducted directly within ATLAS. By reviewing all available structured data in the CDM for a sample of patients satisfying a cohort definition, an adjudicator can classify cases as ‘true positive’, ‘false positive’ (or ‘inconclusive’) in a manner consistent with source record verification.
So, lets review some patient profiles for algorithm #2 in the MarketScan CCAE database!
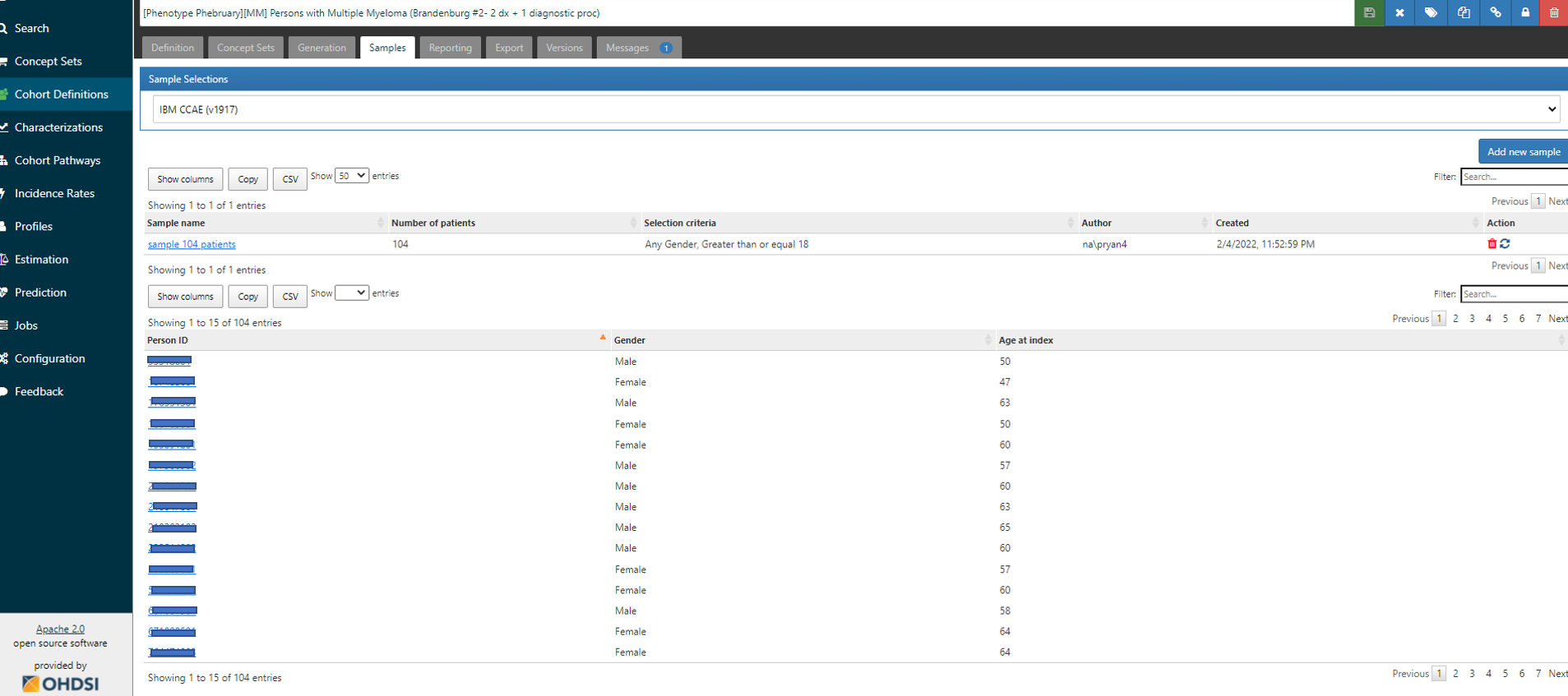
In ATLAS, from the Cohort Definitions left menu, where you’ve built your algorithm under the ‘Definition’ top tab, you’ll note there are other top tabs: Definition, Concept Sets, Generation, Samples, Reporting, Export, Versions, Messages. I start by going to Generation, and executing the cohort definition against the CCAE database (where I find I have 28.151 patients in the cohort). I then toggle over to the Samples tab, and request that I get 104 cases (just like the Optum validation in the Brandenburg study)…
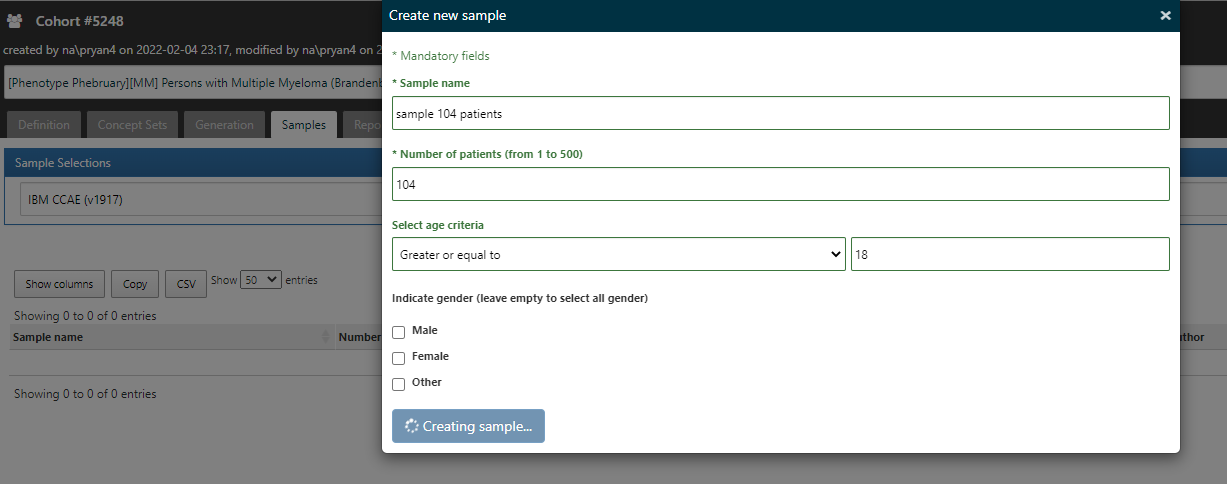
Once the sample is created, I can see links to select any person_id of the sampled cohort members:
Clicking on any of those links brings you to the Profiles tab. You will see a graphical and tabular representation of all events in the persons’ record. The graph plots the events temporally, with day 0 on the x-axis representing the date that the person qualified for the cohort. You can zoom in and out of the timescale by clicking on the graph, and toggle on/off any of the data domains. And you can color specific events of interest. Here, I’m showing you a given person (id removed), zoomed into the year below and two years after cohort entry, restricted to conditions, drugs, measurements, and procedures, with multiple myeloma diagnoses colored in green, diagnostic tests colored in orange, and bone biopsies colored in blue.
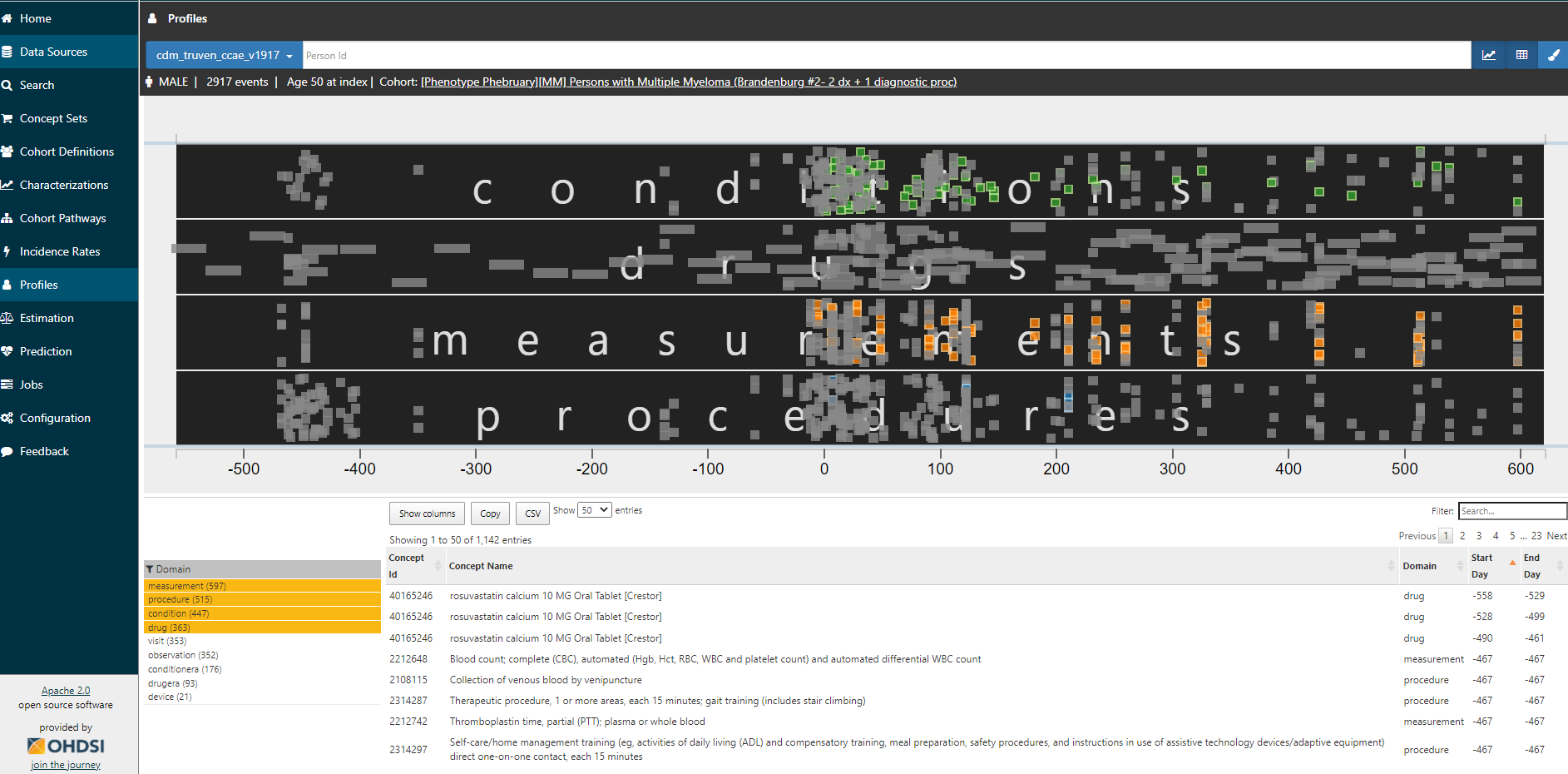
I don’t need to be an oncologist flipping through a paper chart to recognize that this person clearly has multiple myeloma. They have 83 distinct diagnosis codes recorded over two years, more than 100 diagnostic tests, 11 bone biopsy procedure codes. The patient has exposure to various multiple myeloma treatments. including bortezomib (27 drug exposures), ixazomib, dexamethasone, lenalidomide. They take denosumab for bone loss. They are a true positive.
So, tonight, for the fun of it, I reviewed the first 20 of the 104 cases I sampled. ATLAS made it quick and easy, total time was <1 hr. And yet, despite that I’m “only” looking at structured data from an administrative claims database (which often gets disparaged by oncology researchers as being insufficient to study cancers and treatment/regimens), I felt I could make fairly confident conclusions about the case status for almost all of the cases. My strawman tally: 16 ‘confirmed positive’, 3 ‘confirmed negative’, 1 inconclusive. That’d give us a PPV of 84% if we exclude the inconclusive (basically spot on to what Brandenburg observed in Optum). I’ll also note that I found 2 patients with concern about index data misspecification who may have been prevalent cases.
@jon_duke 's team previously presented at OHDSI Symposium about some proposed enhancements to ATLAS to enable formal adjudication of profiles, allowing for reviewers to answer pre-specified questions as they explored the individual cases. When that feature gets released into ATLAS production, I’m sure people will really enjoy using it. But even without it, just the ability to sample cases and review them to draw your own conclusions is extremely helpful. And for those who think chart review is the only way to do validation, this provides a very reasonable approximate that can be directly facilitated by the available data and can be efficiently conducted.
I won’t provide my entire rant here, but validation is quite problematic if all you do is estimate positive predictive value. Without sensitivity and specificity, you can’t truly understand or correct for measurement error. But, given that is very common practice to do chart review and report out a PPV, I hope some will take solace that if you think that’s a valuable aspect of your studies, that you can do it yourself using the OHDSI tools. Knock yourself out.
What do you think can or should be the role of individual case review for phenotype validation? Can electronic patient profiles complement or even possible substitute for source review verification?


 But first, let’s have 28 engaging community discussions to learn from each other. The more that everyone participates and contributes to the conversation, the more that this will become a rich resource that we can all benefit from. Lots of phun Phenotype Phebruary phacts and phigures coming out to showcase important methodological and clinical aspects that are phundamental to all of our OHDSI evidence-generation activities.
But first, let’s have 28 engaging community discussions to learn from each other. The more that everyone participates and contributes to the conversation, the more that this will become a rich resource that we can all benefit from. Lots of phun Phenotype Phebruary phacts and phigures coming out to showcase important methodological and clinical aspects that are phundamental to all of our OHDSI evidence-generation activities.