
Hi folks,
I am Eran from Boston and have been in the immunology and cancer drug discovery space for the past 12 years. I am not a bioinformatician and know very little about relational data bases, though I promise to learn. What I can bring to the table is the cancer researcher perspective and experience. I’d like to thank @Christian_Reich for inviting me to this free global community. Many thanks to all those partaking in the Genomics WG dialogue: Your posts were VERY helpful for me to educate myself and understand what are the main issues and the decisions to be made in these early days. So here is my two cents.
Type of Genetic data
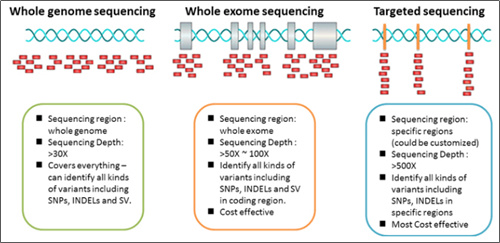
1. Base alterations detected by WGS, WES and real-time PCR:
- Straightforward description of a variant with a single base up to a
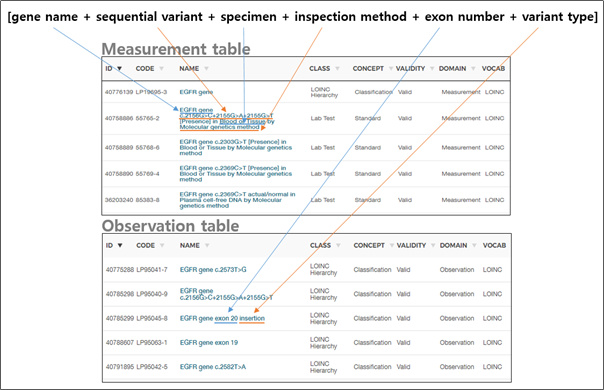
few bases alteration (change, addition, deletion). Cf. ‘Measurement
table’ in slide #7 of presentation 2 by David Fasel. - Need to decide how to handle long insertions, deletions, inversions
as well as multi-variant genes (e.g. Ig, MHC). I think this can be
done consistently. - Unlike other disease cancer is different because it’s mostly about
somatic mutations. In this case it’s important to know thedateof
the biopsy from which genetic information was derived. Chronological alignment with treatment allows queries regarding interplay between therapy, mutation and outcome. - Nomenclature: Consistent, systematic, broad and accommodating
expansion.
2. Gene amplification detected by CGH arrays, NGS, qPCR, FISH, WGS:
- Concept: Gene name, ID, chromosome start/end
- Measure: Copy number
- Value: e.g. 4, ≥6
3. Cytogenetics/ Karyotype, chromosomal aberrations:
- Chromosome number alterations.
- Insertions and Deletions.
- Translocations: e.g. t(9;22)(q34;q11)– CML, ALL (Philadelphia
chromosome). - Rearrangements; Dicentric chromosomes; Fragile sites (germline).
- Reports may be highly variable.
- Ref: International System for Human Cytogenetic Nomenclature (ISCN)
4. Gene expression arrays:
- Compared with mutations and gene amplification these are not hard
data. - Quality of data is affected by samples storage, handling and
processing. - Data obtained using different platforms need to be normalized.
- Large amounts of data and also tissue specific and may change with
time.
5. DNA methylation analysis using arrays or bisulfite with NGS:
- Quality of data might be affected by samples storage and handling.
- Data obtained using different platforms need to be normalized.
Implementation
A. Build stepwise:
- Start with ‘Base alterations’ then move to gene amplifications. Hard
data, high value. - Inclusion of expression and methylation analysis could be discussed
later.
B. Scope- which genetic data to store:
The question is intimately linked to the discussion of flexibility and operationality. One way to ensure efficient querying is to limit observations to variant with clinical interpretation. Another opinion voiced by Kyu Pyo Kim is that all variants should be stored, because variant are not always classified consistently and the scientific knowledge base is expanding. Ergo, additional clinically relevant variants will be discovered.
Let’s not limit OHDSI vision and include all variants. This way OHDSI could become a robust platform for discovery of new prognostic and predictive (i.e. patient stratification) biomarkers. If OHDSI exerts broad impact on human health and spur basic research into the biology of new biomarkers, wouldn’t it be something that we all celebrate?!
It follows that we need to think hard and creatively about the data base architecture/ organization that would support the ‘include all genetic variants’ goal without compromising functionality. As suggested we’ll also have to decide which annotations to include. If push comes to shove and despite best efforts the size of the data base interferes with functionality, we could limit the genetic data base to exome variants (plus promoter regions).
C. Structure of the data base:
As I said bio-computing and data bases are not my cup of tea and I am still educating myself about OMOP CDM. Please be forgiving if my input is flawed. The comparison of the several options in slide 6 of presentation 2 is insightful. Also Kyu Pyo Kim experimenting with a “rigid” and “simple enough” model that keeps the important information and handles large amount of data is instructive.
- If we are to embrace all genetic variants we need to carefully
control what information elements are included per variant. I like
the notion of the essential components as suggested by Kyu Pyo Kim
and shown in David Fasel’s slide 7 of presentation 2. - Annotations: To counteract data burden I tend to support a minimalist
approach, which includes only data items that are required for
friendly, effective and efficient querying. The first seven rows of
the observation table in slide 7 of presentation 2 are an example. It’s up to the community to decide whether other attributes are required for effective querying- ‘population_freq’ (?), ‘interpretation’ (?). All other information could be included in external files if wished so. We should be open to the possibility of discarding certain information if it is deem unnecessary now and in the future. - I understand that there are several ways to organize genetic
information depending on the model: Placing variants in measurement
domain and annotation information in an observation table or in a
genetic data table with a single row per variant. These were a few ideas.
Suggestion:
Treat each variant as a concept with its own ID. It would include the essential components (c.f. first bullet) and a link to the annotation.
- Each variants is generic- a biopsy may have a certain genetic variant
or not regardless of the genetic method used for detection. - Purpose and main advantage: To speed up queries.
- The genetic observation table would consist of a list of IDs of all
variants discovered. If a mutation in a certain gene is absent, it
won’t be on the list. This answers the requirement by Seng Chan You. - Variants observed are linked (foreign key, right?) to the source
specimen. - This requires strict quality check (filtering?) before entries are
accepted into OHDSI CDM because there are no shades of gray- a
variant is either present or not. - Method information (WGS, WES, rt-PCR) and other information
pertaining to run such as quality could be included in an external
file. - Vocabulary is expected to increase by 10^6 (more?) terms.
- Option for hierarchical organization of mutations if found useful. As
example is by chromosome# and position: 1-30,000,000;
30,000,001-60,000,000; 60,000,001-90,000,000 etc.
So this is it for now.
@clairblacketer






 ),
),