
So since we are dong WES, RNA, WGS how can I map these now in OMOP CDM?
@jliddil1 I’m not sure about RNA. We can store the result from WES and WGS, now, though we need to discuss a little bit more and settle the final agreement on it.
So can you point me to some tutorials or info on how to store WEG and WGS?
Wait a second. Scroll up. We are debating what we will have in future, and in which order. And that genomic pipelines starting from the sequences are probably not part of it in V1.
Thanks
I would also like to join.
What is the status of current genomic data model in OMOP? Have we achieved consensus on it? Thanks, -Guoqian
Close, @Guoqian_Jiang. It’s not easy. Please join us at the Genomic meetings. @shilparatwani will invite you.
1 Like
Hello . My name is Mi-So Park from Samsung Medical Center in Korea. For the past two years, in cooperation with Ajou University’s Seojeong Shin (@Seojeong Shin), we conducted a study to apply the results of the genomic test of Samsung Medical Center to G-CDM.
I agree with why table extension is necessary for genomic data, and table expansion is essential.
However, I also agree that the first step is to start with utilizing the currently established CDM tables, rather than applying the four expanded G-CDM tables immediately.
These days we are thinking about the currently established CDM tables to genomic data can be applied and which genomic data can be applied.
Currently, OMOP-CDM is a structure that stores only a few well-known mutations. The ‘Measurement’ table is the best idea to store the genomic data for first step.
What genomic information will be put in the MEASURMENT table?
-
The MEASUREMENT table contains records of Measurement, i.e. structured values (numerical or categorical) obtained through systematic and standardized examination or testing of a Person or Person’s sample.
Figure 1. Description of “Measurement” table -
Because mutation information, the most important measurement data in genomic data, is a structured value obtained by testing person’s sample, it can be applied to the MEASUREMENT table.
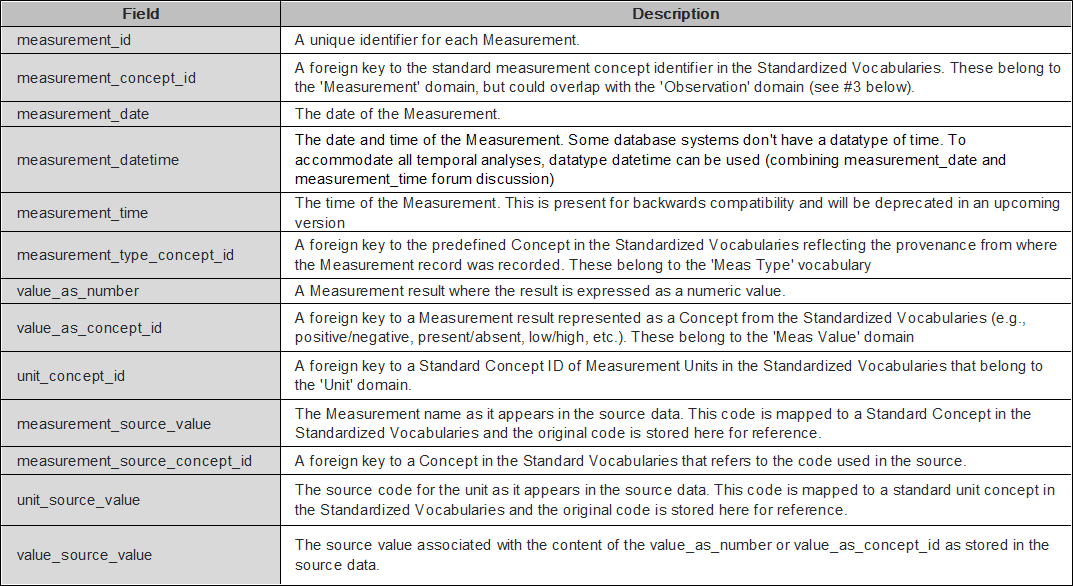
Data type and format of mutation information to be included in the MEASURMENT table
-
Mutation information contains gene symbol, mutation location information, mutation information, etc.
-
To create structure values, we have to take a look at what data to organize and how to use it.
-
Reference sequence (RefSeq) database is a collection of taxonomically diverse, non-redundant and richly annotated sequences representing naturally occurring molecules of DNA, RNA, and protein.
-
Different to the sequence redundancy found in the public sequence repositories, the RefSeq aims to provide a complete set of non-redundant.
-
The non-redundant nature of the RefSeq facilitates database inquiries based on genomic location, or sequence.
Figure 2. Suppressed or redundant RefSeq records -
RefSeqGene, a subset of NCBI’s Reference Sequence (RefSeq) project, defines genomic sequences to be used as reference standards for well-characterized genes and is part of the LRG Project.
-
RefSeqGene be used as a stable foundation for reporting mutations, for establishing conventions for numbering exons and introns, and for defining the coordinates of other biologically significant variation.
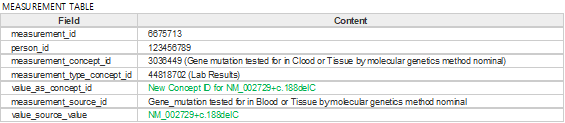
Figure 3. Example of measurement table with variant information applied
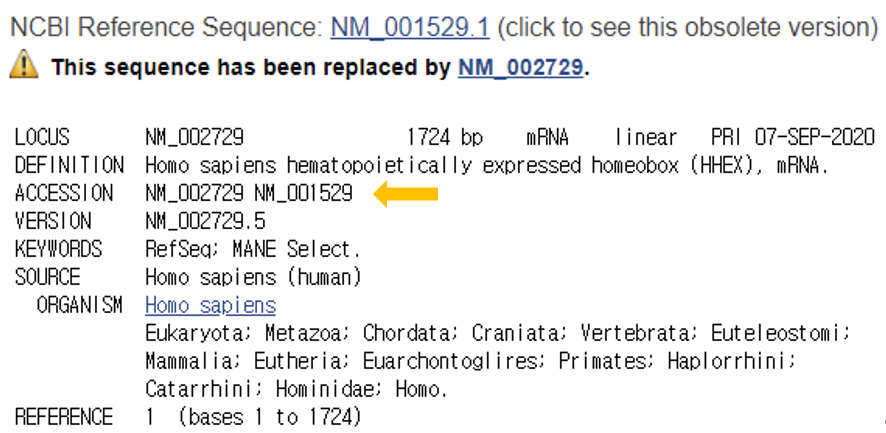
Another proposition (LRG project)
-
LRG (Locus Reference genomic) is specifically created for the reporting of clinically relevant variants and hence, are for loci with clinical implications.
-
LRG are stable and therefor are not versioned, thus reducing ambiguity when reporting variants.
-
When an LRG is established for any gene, the RefSeq Gene and its annotation will be frozen to match that of the LRG
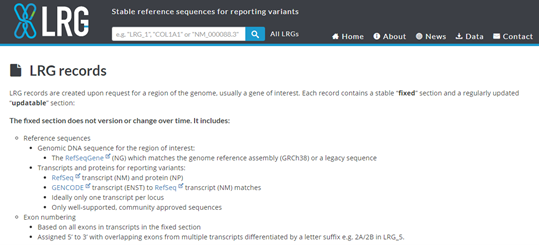
Figure 4. LRG records report -
Versioning is an issue with traditional reference sequence records simply because the actual sequences differ from version to version for records with the same accession number.
-
A variant description such as LRG_13:g.8290C>A will always remain valid and will never be subject to misinterpretation.
-
Over 1283 LRGs have been created, of which 862 are public.
-
That is why the user simply needs to ensure that the LRG contains all of the necessary transcripts for the intended task.
To applied genomic information to current OMOP-CDM, we proposed several methods that how to show mutation.
What is certain is that need on provide both RefSeq (or LRG) and mutation information, not gene symbols.
What do you think of Which of RefSeq and LRG can efficiently represent the variants, or any suggestions for another good method?
We look forward to your opinions. Thank you.
1 Like
Very timely message, and we are clearly behind in disseminating what the Oncology WG has done. It will all be visible at the Symposium.
But take a look at Athena and the latest vocabulary release. It essentially did what you are suggesting:
- All genomic concepts are domain_id=‘Measurement’
- We incorporated the HGNC canonical human genes (vocabulary_id=‘HGNC’), but declared them as variants of those genes (as the intact gene is not a finding).
- We built genomic variants based on a number of collections (Jax, Clinvar, CIViC, cgi, CAP, NCIt), instead of the totality of all possible variants. We are working with other collections (oncoKB and Cosmic).
- If the variant is defined at the molecular level the HGVS notation is the concept_code, with the reference sequence provided by the source vocabularies. These are on the genomic, transcript or protein levels
- Otherwise there are less precise variants (e.g. Protein expressions)
Start for example with this and click yourself through: https://athena.ohdsi.org/search-terms/terms/35955862
Thoughts?
2 Likes
How can I join the Genomic WG?
Hi Nicole, My name is Shilpa Ratwani and I coordinate the Genomic WG efforts. We have a weekly call every Tuesday at 9:00AM EST where the group meets to discuss the activities related to the Genomic Model and Vocabulary. I will send you and invite to that meeting which will include the conference line information.
We are currently preparing for the OHDSI 2020 Symposium as we will be showcasing the work we have completed so far in the Genomic space. We have made great progress this year and have successfully loaded version 1 of the variant database into the OMOP Vocabulary. We are in the process of validating the Vocabulary against the partner variant lists and also finalizing the Genomic Model.
We are looking forward to your participation in the upcoming Genomic WG discussions.
hello, @Shilpa_Ratwani,
We are very interested in this work (I work for FinnGen, having >250k dna samples).
When will this be showcased ??
I can’t find it on the Symposium’s program.
Thanks
Hi all! I’d be interested in using the G-CDM for some targeted NGS data. I’ve been browsing the drive to get the specifications but I am not sure the excel in the 1.Specification folder is the most updated version as the attributes on the tables do not match those available in the presentation. What I missed the most was the start and end position in the target_gene table as well as the updates in the variant_annotation one.
Has a candidate version been release somewhere?
Many thanks!
Hi, Ana. I’m Seojeong at Ajou University, Korea.
The OHDSI Genomic working group has been discussing the way to store genomic data into the Measurement table of OMOP-CDM. More recently, we have been working on vocabulary as you can see in @Christian_Reich 's post.
If you still are interested the old version of genomic-cdm, you can refer this link. On that site, the table specification is the latest version rather than the ERD.png which is in the README page. As you noticed, in the TARGET_GENE table, position information is not presented anymore because they were considered not necessary. Likewise, the VARIANT_ANNOTATION table specification is also not synchronized with ERD.png.
Under the consideration of the recent direction of the working group, I recommend you to adopt the MEASUREMENT table in order to store NGS data. But if you want to store variant with metadata, you can use the older version of genomic-cdm with 4 tables.
2 Likes
Hi @1118! Many thanks for your help. I see there should a link in “the way” but it is not working for me.
How would we be able to captures the genes targeted by different gene panels and compare variants obtained through them?
Also is the idea to relate the annotations, VAF, coverage, and other information associated to the variants through fact_relationships?
Apologies if all that information is available somewhere and I have not been able to find it.
By mapping them to one of the new Genomic Marker concepts and placing them into the MEASUREMENT table. We are in the process of documenting what how that works. But bottom line: There is a hierarchy with increasing level of molecular detail.
We don’t have a way to represent the methods of obtaining genomic information. So, whether it’s panels (microarray or PCR) or sequencing - we don’t distinguish it right now. And we are not sure we need to.
None of it. That is the job of the sequencing pipelines. There are public and commercial solutions for this. We don’t want to invent another wheel. Somebody needs to call a genomic variant, and then we take it, give it a Concept representation and put it into MEASUREMENT. This will allow all the downstream use cases of figuring out the effect of variants on prognosis and treatment outcome. That’s were we want to be.
Please join us in the Genomic/Onco WG.
1 Like
How do I join the Genomic/Onco WG? I work with CODATA in East Africa and we are in the process of growing an OMOP implementation with the G-CDM extensions.
Thanks