Just went through Drive documents. I admit it is an ambitious goal to put genomic data to structured database model not only because of the data size, but also because of the non-standardized notation used for genomic data. However, there is still a strong need for a good database format for that.
We have just calculated pharmacogenomics recommendations for 44,000 biobank participants (https://www.nature.com/articles/s41436-018-0337-5) and I only wanted to highlight that information about genotype PHASING (are the genotypes of 2 SNPs phased) and IMPUTATION (was the genotype imputed, what was the value probability) are also very important pieces of information when you are dealing with genomic data.
Thank you for your information about “Phasing” and “Imputation” for dealing sequencing data.
I’ll review that issue and let G-CDM adopt those concepts in adequate position and method!
Thanks again
Hello. Is [CDM Builders] the best place to follow the oncology/genomics CDM extension discussions, or are there different discussion groups for those topics? Thank you.
First of all, I appreciate for all your interest and discussion to support the genomic CDM Workgroup.
These days we are thinking basic concepts; why we need (do we need) a table extension for genomic data; which elements of genomic data has to be stored in OMOP-CDM; how the genomic CDM can be used in clinical practice.
Why is genetic material important in the clinical practice?
The patient’s genomic data are used as an indicator for determining the cancer stage and anticancer treatment according to the NCCN guidelines (Figure 1).
Figure 1. Genomic alteration data used in clinical decision
The way to store the genomic data in current OMOP-CDM
Currently, OMOP-CDM is a structure that stores only a few well-known mutations.
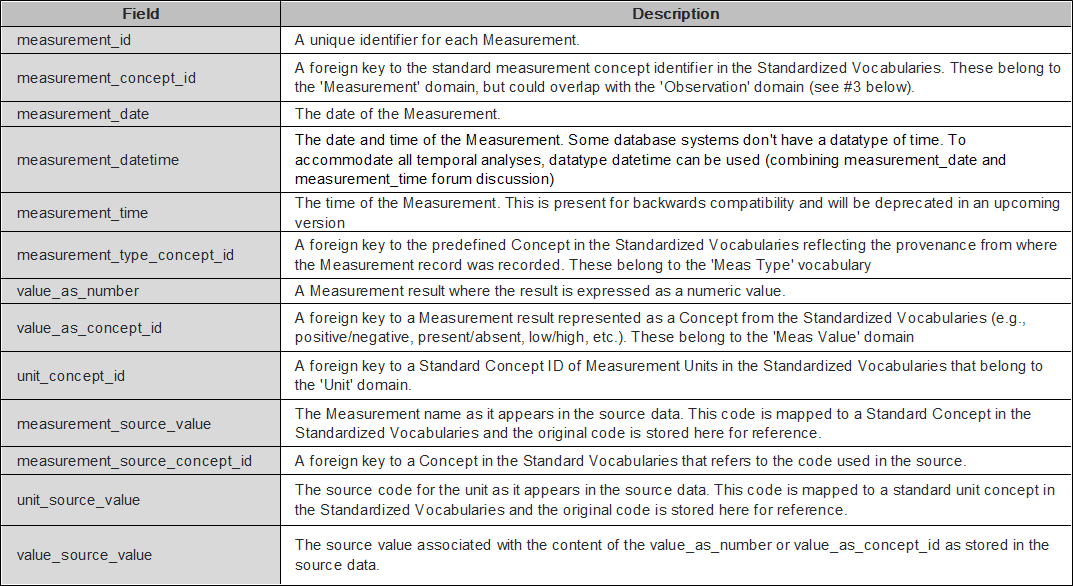
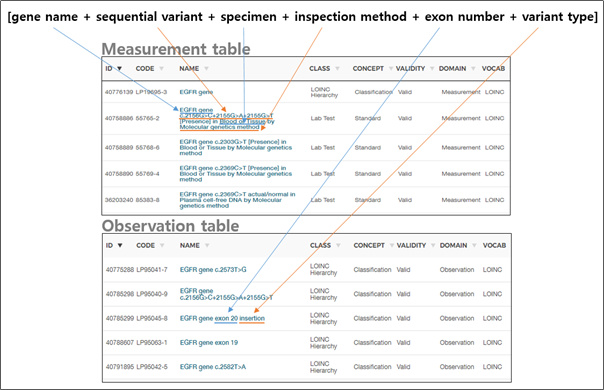
In the Measurement/Observation table, the combination of [gene name + sequential variant + specimen + inspection method + exon number + variant type] information is created in one set and has a concept ID (Figure 2).
Figure 2. How to present a variant in the current OMOP-CDM
This is an efficient way to store the result of only a few typical mutations, such as a single gene or dozens/hundreds of specific mutations, to express whether or not those mutations are present.
Why does OMOP-CDM need to be expanded?
Reason 1: The number of variant subject to sequencing has increased exponentially.
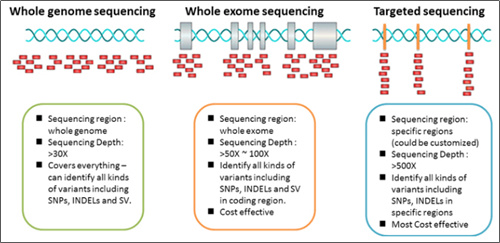
For the recently generalized Targeted Next Generation Sequencing (Targeted-NGS) technology, the number of genes to be examined is among tens and hundreds, and each gene has its own area for testing.
However, countless variations can occur at a sequence (A), such as deletions (A to -), substitution (A to T / C / G), and insertions (A to AT / ATG / ATGC / ATGCC / ATGCCTTACGGAT and so on……).
It is impossible to make the number of all these cases into one set, as it is now.
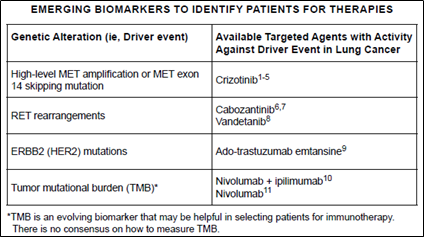
In particular, Whole Exome Sequencing (WES) and Whole Genome Sequencing (WGS) technologies examine entire exome and genome sequences, respectively, and are being used to find patients who can be treated with certain drugs as it has been found that Tumor Mutation Burden (TMB)* tests in immune diseases are related to predicting Immune checkpoint inhibitor (ICI) treatment effects (Figure 4). (DOI: 10.1200/JCO.2017.75.3384 Journal of Clinical Oncology 36, no. 7 (March 1 2018) 633-641.)
Figure 4. TMB as an emerging biomarker in NCCN Guidelines Version 1.2019 Non-Small Cell Lung Cancer
*TMB (Tumor Mutation Burden): TMB is the calculation of the number of mutations in cancer cells.
The higher the TMB, the higher the response to immunosuppression drugs is shown.
Because WES and WGS technologies examine the entire exome and genome sequence, it is inefficient to create all the variation concepts as they are today, as with targeted-NGS.|
Reason 2. Non-variant information is also required to interpret NGS testing.
In addition to the current measurement/observation table, there are features that should be taken into account when comparing multi-center data of NGS testing.
This is because the method of the NGS testing is not standardized like other clinical lab test tests.
They should be recorded/standardized together to interpret NGS testing between institutions to further classify the patient’s genetic variation and cancer condition and to identify the outcome of clinical care.
Currently, most of these data may be retrieved from pathology reports or from EMR, even though it’s very difficult, but in CDM, there is no way to obtain the well refined NGS results.
> Non-vairant features to be recorded/standardized
Sequencing platform (device & software) information
: Information about the name and version of the platform using sequencing assigned by the institution.
Reference genome
: Prior knowledge that is used in aligning the leads to recognize changes in sequence. Because the reference genome acts as a comparison criterion, it is important to confirm whether the reference genome is identical or not when comparing variants between institutions.
Read depth (variant & total)
: Reads are the thousands of pieces of nucleic acid that you analyze when you run an NGS. NGS amplifies the reads and sequencing them over and over again, supplementing slightly incorrect parts to increase the accuracy. Read depth is the number of times a single location has been read during sequencing. Bigger read depth means better accuracy. You should exclude variants with read depth under threshold during analysis for increasing accuracy of comparison.
Genotype
: Whether the variant comes from somatic or germline
Annotation information
: The clinical impact of the variant
Things you can do with Genomic-CDM (Use cases)
Identifying patients for therapies
: By using variants data resulted from NGS, you can figure out how many patients are adequate for immunotherapy (ex. Nivolumab) in each centers by calculating tumor mutational burden (TMB) from the result data of whole exome sequencing (WES).
Selecting patients for clinical trials
: By using variants data and linked clinical data, you can find and encourage patients to involve certain clinical trials (ex. NCT02296125) who have specific mutation profiles and treatment history.
Finding genes/variants related to outcomes
: By using variants characteristic and linked drug/treatment exposure data, you can discover genomic characteristic of patient group having a poor reaction to drugs.
You can also use non-variant information of the genomic-CDM in…
confirming the comparability of the genomic data between institutions by using information of sequencing platform, reference genome, genotype.
filtering the genomic data by quality by using information of read depth of variant and total target read.
The meeting of the genomic CDM working group has been suspended some while, but we will hold the meeting again soon. We look forward to your participation.
That is very good. Thanks for putting new life into it.
Because we do need to get to the bottom of this problem. So far, we have mostly collected a pile of potential pieces of information, which could be useful. The problem with that approach is that we may spin and spin with little results:
We have no standard model
We have not enabled network research
We have not enabled or developed standardized, systematic or scaling methods
As a result, we are still far away from generating scientific insights.
The only chance I see is to work this from the top down:
Start with the use cases
This is a good starting point. But we need to agree on them. Even if we have a longer list with high and low priority it would help tremendously.
Personally, I would split the last one into
Utilize established genes/variants in estimation/prediction/diagnostic models
Establish (explore and identify) genes/variants for estimation/prediction/diagnostic purposes
We also need to declare what we are not going to tackle. For example, building a genomic pipeline from raw data.
Once we have these, we continue with
Building the model
What data about patients do we need for these use cases, and how do we represent them. There is good work available from @ShinSeojeong, @1118 (we should not have two concepts for the same person, btw, violation of OMOP CDM ), @KKP1122 and @Yurang_Park. But we need to come up with a compact and efficient solution. Not all data, just because they exist, should be standardized. Only if they serve the use cases.
We need to outsource all prior knowledge, like known variants and their relevance, into the vocabularies, and take it out of the data
We need to standardize the representation of the data in the vocabularies
This should be an iterative process. V1 should the minimal model to make progress against the use cases. Once we have that, we need to:
Put some data into V1 Test them against use cases Expose the results to the community
Before that, we should not go into V2.
Another rule we have successfully applied to OMOP CDM and Vocabulary development is the notion of “theft”. We should not invent anything that exists and is established. We should evaluate existing systems and adopt, instead of creating new ones. That will require some work. I can help with vocabularies. @1118’s mention of the NCCN is a first good step.
@jliddil1 I’m not sure about RNA. We can store the result from WES and WGS, now, though we need to discuss a little bit more and settle the final agreement on it.
Wait a second. Scroll up. We are debating what we will have in future, and in which order. And that genomic pipelines starting from the sequences are probably not part of it in V1.
Hello . My name is Mi-So Park from Samsung Medical Center in Korea. For the past two years, in cooperation with Ajou University’s Seojeong Shin (@Seojeong Shin), we conducted a study to apply the results of the genomic test of Samsung Medical Center to G-CDM.
I agree with why table extension is necessary for genomic data, and table expansion is essential.
However, I also agree that the first step is to start with utilizing the currently established CDM tables, rather than applying the four expanded G-CDM tables immediately.
These days we are thinking about the currently established CDM tables to genomic data can be applied and which genomic data can be applied.
Currently, OMOP-CDM is a structure that stores only a few well-known mutations. The ‘Measurement’ table is the best idea to store the genomic data for first step.
What genomic information will be put in the MEASURMENT table?
The MEASUREMENT table contains records of Measurement, i.e. structured values (numerical or categorical) obtained through systematic and standardized examination or testing of a Person or Person’s sample.
Because mutation information, the most important measurement data in genomic data, is a structured value obtained by testing person’s sample, it can be applied to the MEASUREMENT table.
Data type and format of mutation information to be included in the MEASURMENT table
Mutation information contains gene symbol, mutation location information, mutation information, etc.
To create structure values, we have to take a look at what data to organize and how to use it.
Reference sequence (RefSeq) database is a collection of taxonomically diverse, non-redundant and richly annotated sequences representing naturally occurring molecules of DNA, RNA, and protein.
Different to the sequence redundancy found in the public sequence repositories, the RefSeq aims to provide a complete set of non-redundant.
The non-redundant nature of the RefSeq facilitates database inquiries based on genomic location, or sequence.
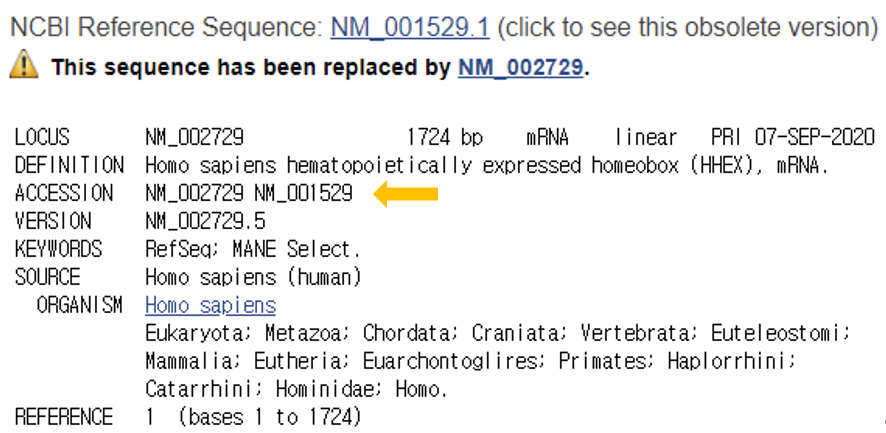
RefSeqGene, a subset of NCBI’s Reference Sequence (RefSeq) project, defines genomic sequences to be used as reference standards for well-characterized genes and is part of the LRG Project.
RefSeqGene be used as a stable foundation for reporting mutations, for establishing conventions for numbering exons and introns, and for defining the coordinates of other biologically significant variation.
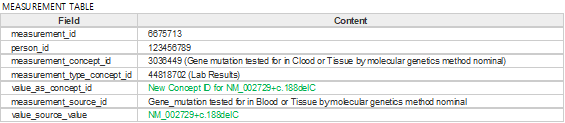
Figure 3. Example of measurement table with variant information applied
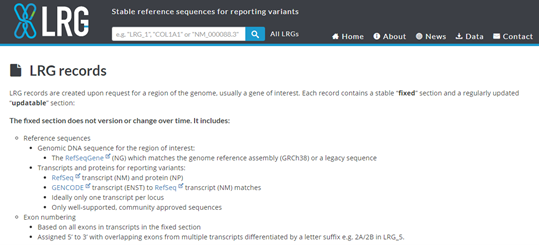
Another proposition (LRG project)
LRG (Locus Reference genomic) is specifically created for the reporting of clinically relevant variants and hence, are for loci with clinical implications.
LRG are stable and therefor are not versioned, thus reducing ambiguity when reporting variants.
When an LRG is established for any gene, the RefSeq Gene and its annotation will be frozen to match that of the LRG
Figure 4. LRG records report
Versioning is an issue with traditional reference sequence records simply because the actual sequences differ from version to version for records with the same accession number.
A variant description such as LRG_13:g.8290C>A will always remain valid and will never be subject to misinterpretation.
Over 1283 LRGs have been created, of which 862 are public.
That is why the user simply needs to ensure that the LRG contains all of the necessary transcripts for the intended task.
To applied genomic information to current OMOP-CDM, we proposed several methods that how to show mutation.
What is certain is that need on provide both RefSeq (or LRG) and mutation information, not gene symbols.
What do you think of Which of RefSeq and LRG can efficiently represent the variants, or any suggestions for another good method?
We look forward to your opinions. Thank you.
Very timely message, and we are clearly behind in disseminating what the Oncology WG has done. It will all be visible at the Symposium.
But take a look at Athena and the latest vocabulary release. It essentially did what you are suggesting:
All genomic concepts are domain_id=‘Measurement’
We incorporated the HGNC canonical human genes (vocabulary_id=‘HGNC’), but declared them as variants of those genes (as the intact gene is not a finding).
We built genomic variants based on a number of collections (Jax, Clinvar, CIViC, cgi, CAP, NCIt), instead of the totality of all possible variants. We are working with other collections (oncoKB and Cosmic).
If the variant is defined at the molecular level the HGVS notation is the concept_code, with the reference sequence provided by the source vocabularies. These are on the genomic, transcript or protein levels
Otherwise there are less precise variants (e.g. Protein expressions)





 ),
),