If you thought you were going to get out of PHebruary, without PH, well, you haven’t been paying attention. Without further ado I introduce to the reader: Pulmonary Hypertension.
I’ve reached out to and involved a friend and colleague, Jason Weatherald (not yet on the forums but joining soon). Some of you might recognize his name, we’ve been working together with Patrick Alba, Scott Duvall, and others at the VA, as well as Jose Posada on the Covid Prone NLP study. Jason is a pulmonologist who did his fellowship in pulmonary hypertension, he’s just relocated to the U of Alberta (in Edmonton) from Calgary, although we won’t hold that against him.
Frankly, for many of these descriptions, I feel like I should have received CME credit for reading them (looking at you in particular, @Allen Wu). As an internist, I vowed to love all my children organ systems equally, however I do think the gauntlet has been thrown down to convince the audience that the heart and lungs are at least as cool as the brain.
There’s another method to this madness, in that Jason and I have been discussing and think there’s exceptional alignment between the capacities and interest of the OHDSI community, and the RWE research needs within pulmonary HTN, so part of this is to generate some interest in a really interesting area of pathophysiology and emerging therapeutics, and lead some work in this area.
We’ll start with the clinical description, and follow that up with a post later on regarding phenotyping background and efforts to date.
Pulmonary Hypertension Clinical Description
Background
A reasonable approximation for the expected pressures in the heart and downstream arteries / capillary networks, and handy memory aid, is the ‘rule of 5’s’, illustrated below (numbers represent pressure in mmHg)
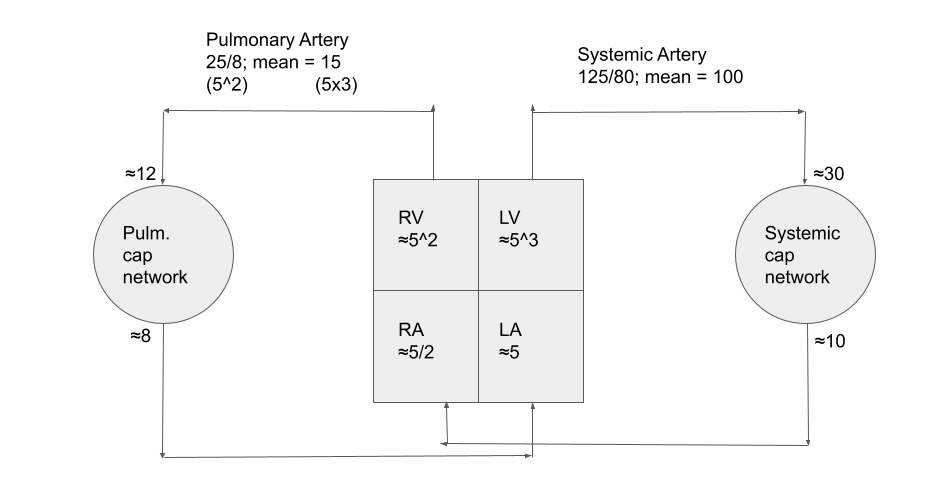
Flow within the pulm cap network is determined through the competition between hydrostatic forces (i.e. perfusion pressure + gravity) and atmospheric pressure: they are effectively a network of Starling resistors. In states of airspace disease, the arterioles (i.e. just upstream of the air units) constrict in response to low partial pressure of oxygen. In this way, blood perfusion is preferentially directed to the healthy areas of the lung (referred to as ventilation:perfusion matching). It also intuitively explains why states of chronic hypoxemia can lead to pulmonary hypertension over time.
Interestingly, and perhaps to make a point, the lungs do not need the brain or nervous system to do this ![]() ; excised lungs regulate their blood flow just fine.
; excised lungs regulate their blood flow just fine.
Technically the definition of pulmonary hypertension is a mean pulmonary artery pressure of 20mmHg (recently revised downward from 25mmHg), and is 2SD above normal. The only way to directly measure the PAP is through a right heart catheterization (i.e. floating a balloon catheter from a vein in your neck, through the right atrium, down to the RV, and into the pulmonary artery). Doing so allows a set of pressure measurements in the cardiac chambers. Advancing the catheter further until the balloon wedges into a small branch of the PA effectively creates a static fluid column from which we can infer whether pressures are high in the LA (and the LV, at the end of diastole). We call that the pulmonary artery wedge pressure (PAWP).
In practice, that procedure is typically used for a small subset of PH patients, suspected to have pulmonary arterial hypertension.
Etiology & Pathogenesis
The two main frameworks are oft used to split up etiologic considerations of PH. Most common is to consider the WHO’s classification of PH. These include:
Group 1 – Pulmonary arterial hypertension (PAH)
Group 2 – PH due to left heart disease
Group 3 – PH due to chronic lung disease and/or hypoxemia
Group 4 – PH due to pulmonary artery obstructions
Group 5 – PH due to unclear multifactorial mechanisms
Another is to consider hemodynamic profiles that are determined at RHC: pre capillary disease (group 1, 3, 4 and sometimes 5), post capillary disease (group 2 and group 5), combined pre and post capillary PH (group 2 and 5), and high cardiac output states, which connect nicely to the physics of blood flow.
I’ve included a figure that ties pathogenesis to clinical findings. The broader project (which tends to focus on that connection) is one that the U of Calgary medical school has driven forward as a theme of its teaching for some time. It has picked up enough steam to get translated into various languages. The content is free for use under the Creative Commons license - so feel free to consult and incorporate the images in ‘clinical definition’ phase of any phenotyping work, can can be freely posted.
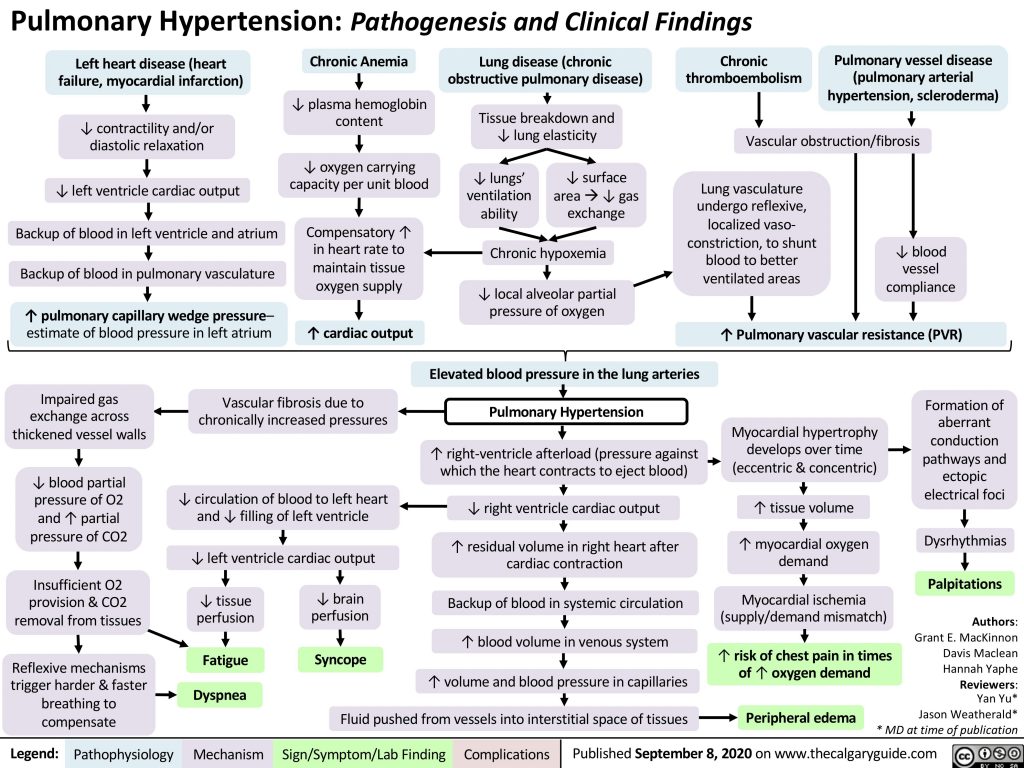
The mechanism boxes include the physiologic adaptations that occur over time.
Diagnosis
Epidemiology: overall PH estimates range from a prevalence of 1-9% (CCS Guidelines). Group 2 PH is the most common, followed by group 3.
Symptoms and signs: shown above in green
Initial investigations in patients with that set of symptoms include basic labs, EKG, often a CXR and an echo.
In practice, the echo is often the first piece of hard evidence for PH, through estimates of RV systolic pressure. This is done by looking at the velocity of regurgitant flow across the Tricuspid valve (ie. from RV to RA in the figure), and making assumptions about RA pressure. The RV systolic pressure can then be estimated from the jet velocity. This estimation can be inaccurate, especially when someone is otherwise unwell (which they usually are, when the index echo is in the hospital). As a result, it’s reasonable to get a confirmatory echo well out from the acute phase of illness. An echo is inadequate to confirm a diagnosis of PH; RHC is needed. However, echo can be highly suggestive especially when the right cardiac chambers are dilated.
A series of other surrogates on echo related to RV and valve morphology and movement are also used. An echo can also find many features that would be suggestive of left heart disease, suggesting a group 2 PH.
An EKG can also demonstrate features of increased RV mass, by changing the net depolarization vector of the ventricles (manifests in the QRS complex to give Right Axis Deviation (RAD, in red) and a positive R in V1 (purple in figure) and atria (effectively increasing the AUC of the P wave in lead II, not evident in this ECG); as well as some repolarization changes (blue).
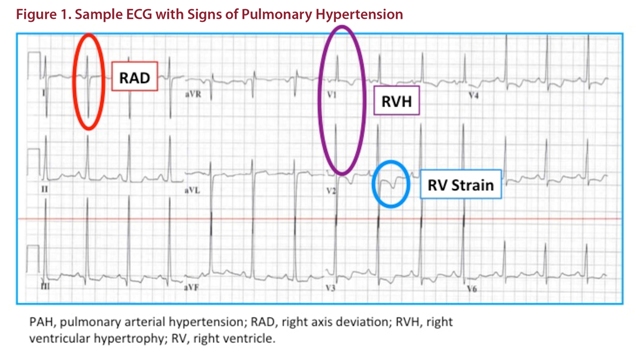
Usually, if one sees an echo estimated RVSP > 50mmHg (in acute illness) a component of PH is suspected, assuming that it takes some RV remodeling to generate pressures higher than that, even if the person is unwell.
After an echo, depending on clinical suspicion, diagnostic refinement may include ABG, ANA / ENA / RF / Anti-CCP / Anti Scl-70, HIV, TSH; CTPE / V-Q scan/ High Res CT / PFT / Sleep Study; all of which look for a host of secondary causes.
Because it is invasive, right heart catheterization (RHC) is often reserved for patients suspected to have Group I (Pulmonary Arterial Hypertension PAH) or Group IV (Chronic thrombotic pulmonary hypertension, CTEPH), or for cases of diagnostic uncertainty as to the contribution of PAH. This can be health system / incentive dependent. During RHC, fluid challenge / vasodilator testing can be performed to clarify diagnosis and to assess for potential response to therapies.
Group I PH, i.e. PAH, is confirmed by a RHC showing mean PAP >20, Wedge pressure <15, and Pulmonary Vascular resistance (derived using a hydrologic analogy to Ohm’s law: ~ ( mPAP - mean PAWP)/CardiacOutput)) >3 Wood units.
The distinction between precapillary and post capillary causes is not perfect, even with RHC, as shown in the figure below. Ideally, you’d see all the blue dots below the horizontal dotted line of PAWP of 15mmHg - so there is still risk of misclassifying patients even with RHC. It’s clear that molecular diagnostics would be a major boon for the area.
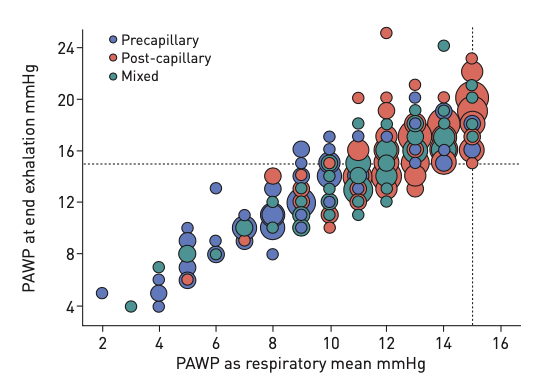
(From Rosenkranz S, Preston IR. Right heart catheterisation: best practice and pitfalls in pulmonary hypertension. Eur Respir Rev. 2015;24:642-652)
Therapy
Many of the WHO groups are managed by trying to treating the underlying condition. That is a large set of diseases, so we will focus on Group I (PAH).
As a quick summary, some therapeutic avenues include:
- Therapeutic anticoagulation in select patients (with idiopathic, heritable, or drug / toxin induced PAH).
- Calcium channel blockers (in the same subset, when hemodynamic testing shows marked short term improvement with an inhaled vasodilator such as nitric oxide).
Agents that target PAH:
- Prostacyclin pathway. Patients with PAH have deficient levels of prostacyclin. Epoprostenol (IV), Treprostinil (IV, SC, PO or inhaled), Iloprost and Selexipag are examples of agents on this pathway.
- Endothelin receptor antagonists (ERA). PAH patients have elevated levels of endothelin-1, a potent vasoconstrictor. Three ERAs are available: macitentan, ambrisentan, bosentan.
- NO-cyclic GMP. PAH patients are deficient in endogenous NO. This can be treated with phosphodiesterase type-5 inhibitors (PDE-5i), or soluble guanylate-cyclase stimulators.
It is noteworthy that some patients with group II (Left heart disease) PH may undergo a therapeutic trial (typically < 3 months(?)) of a single agent (typically PDE-5i) in select cases.
There is recent data and FDA approval for inhaled treprostinil (Tyvaso) in group 3 PH due to interstitial lung disease.
Upfront dual oral combination therapy is now recommended in most patients with PAH. The most severe high-risk patients are treated with parenteral drugs like IV epoprostenol or treprostinal IV/SC, often in combination with two oral drugs.
The treatment of choice for CTEPH is a surgical procedure called pulmonary thromboendarterectomy which removes fibrotic material from the inside of the PAs. Other treatment options include riociguat (a soluble guanylate-cyclase stimulator) and/or off-label use of the other PAH therapies above and balloon pulmonary angioplasty.
Prognosis
The prognosis of PAH is poor. Median survival was 2.8 years in the 1980s before the above therapies were discovered and tested. In modern registries, the median survival has improved but remains poor at 5-7 years, depending on the cause and severity.
The prognosis for CTEPH depends largely on whether patients can be treated with pulmonary thromboendarterectomy. Eligibility for this procedure depends on technical factors (location of obstructive thrombo-fibrotic material in the pulmonary arteries) and patient factors (age, comorbid conditions). For patients who undergo surgery successfully, 5-year survival is >90%.
Prognosis among other classes depends on the underlying disease, but invariably remodeling your RV in response to any disease process reflects a high burden of illness.
Next Steps
Next post will focus on review of phenotyping efforts; the terminology space, and preliminary work.