We have been allowed to submit a bonus phenotype for this Phenotype Phebruary. We would be hugely helped if the OHDSI community can help evaluating our phenotype. Evaluation is due on February 15th 2023.
We present here the Clinical Description, Literature research and Atlas Cohort Definitions for NF1 and NF1 with OPG. This effort is coming from EU-PEARL, a collaboration between academia and EFPIA around platform trials funded by Innovative Health Initiative. Neurofibromatosis (NF) is one of the selected disease areas. A validated, computable phenotype for NF would help in trial site selection through accurately retrieving patient counts. Within NF, we are specifically interested in developing a phenotype for Optical Pathway Glioma (OPG) as manifestation of NF is often not captured in detail in health records.
I am posting this on behalf of Britt Dhaenens (Erasmus MC), Cheryl Tan (Uni of Oxford) and Susana Kalko (Vall D’Hebron).
Clinical Description
Overview: Under the term Neurofibromatosis is (NF) we distinguish three closely related disorders: Neurofibromatosis type 1 (NF1), Neurofibromatosis Type 2 (NF2) and Schwannomatosis (SWN). NF1 is a hereditary disorder that predisposes to the development of nerve sheath tumours. These tumours are mostly benign with a low chance of malignant transformation, but can cause significant neurological morbidity due to their size and/or location. It has an incidence of approximately 1 in 2500 births. The disease is autosomal dominantly inherited, with a high de novo mutation rate of 50% (50% of the patients with NF1 have an affected parent). NF2 is associated with the development of bilateral Vestibular Schwannoma (VS), which result in hearing loss. SWN is very rare, and results in the development of painful schwannomas throughout the body.
Optic pathway glioma (OPG) is the most common tumour in NF1, but can occur in patients without NF1 as well. Approximately 15-20% of children with NF1 will develop an OPG. These are mostly benign tumours that are located in the optic pathway in the brain.
Presentation: NF1 is a neurocutaneous disorder: most of the disease manifestations are located in the skin and the nervous system, but NF1 shows disease manifestations in nearly all organ systems. Manifestations can develop from childhood into adulthood. Patients are characterized by café-au-lait macules (coffee with milk-coloured spots on the skin) and freckling in the inguinal region. Adults also develop cutaneous neurofibromas. During the course of the disease, they can develop a variety of tumours, including plexiform neurofibromas and tumours of the brain (low grade brain glioma, including optic pathway glioma, and high grade brain glioma). Additionally, they have an increased risk for other cancers (e.g. breast cancer). Cognitive impairment, and cardiovascular and musculoskeletal abnormalities may also occur, such as hypertension and scoliosis. NF1 is characterised by a prominent variability in the prevalence, timing and severity of disease manifestations.
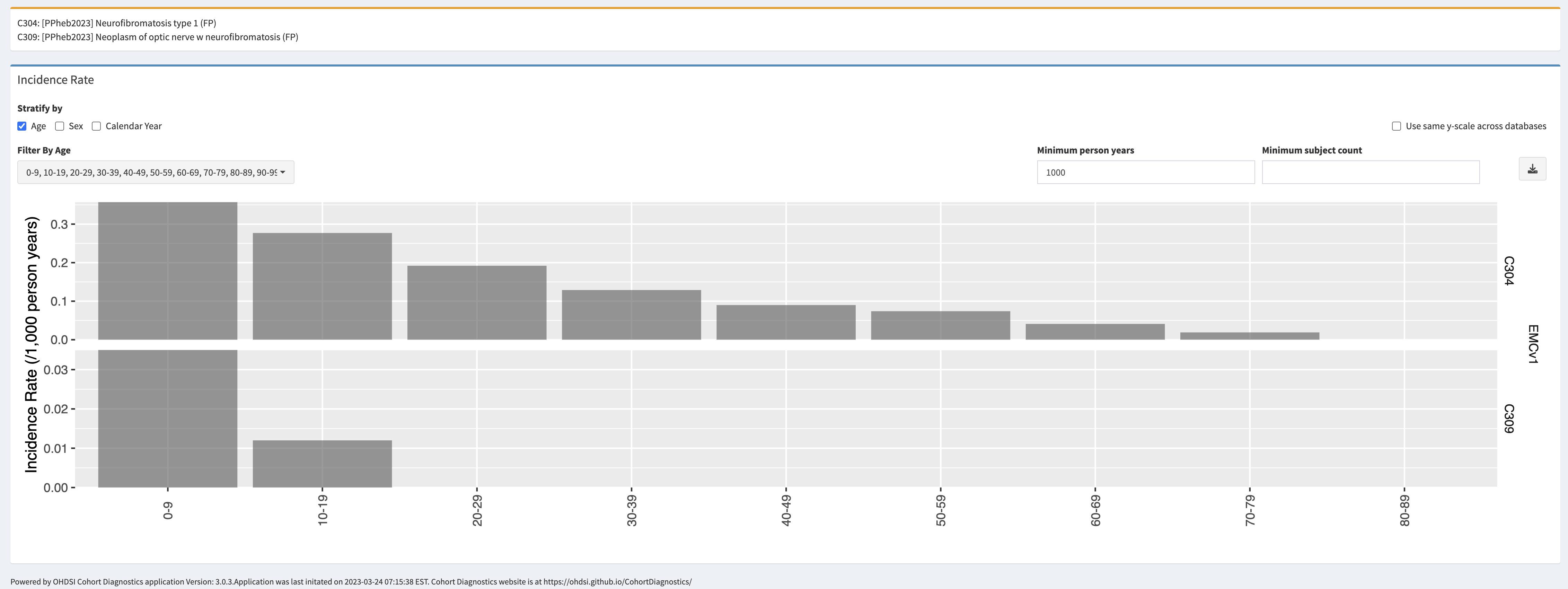
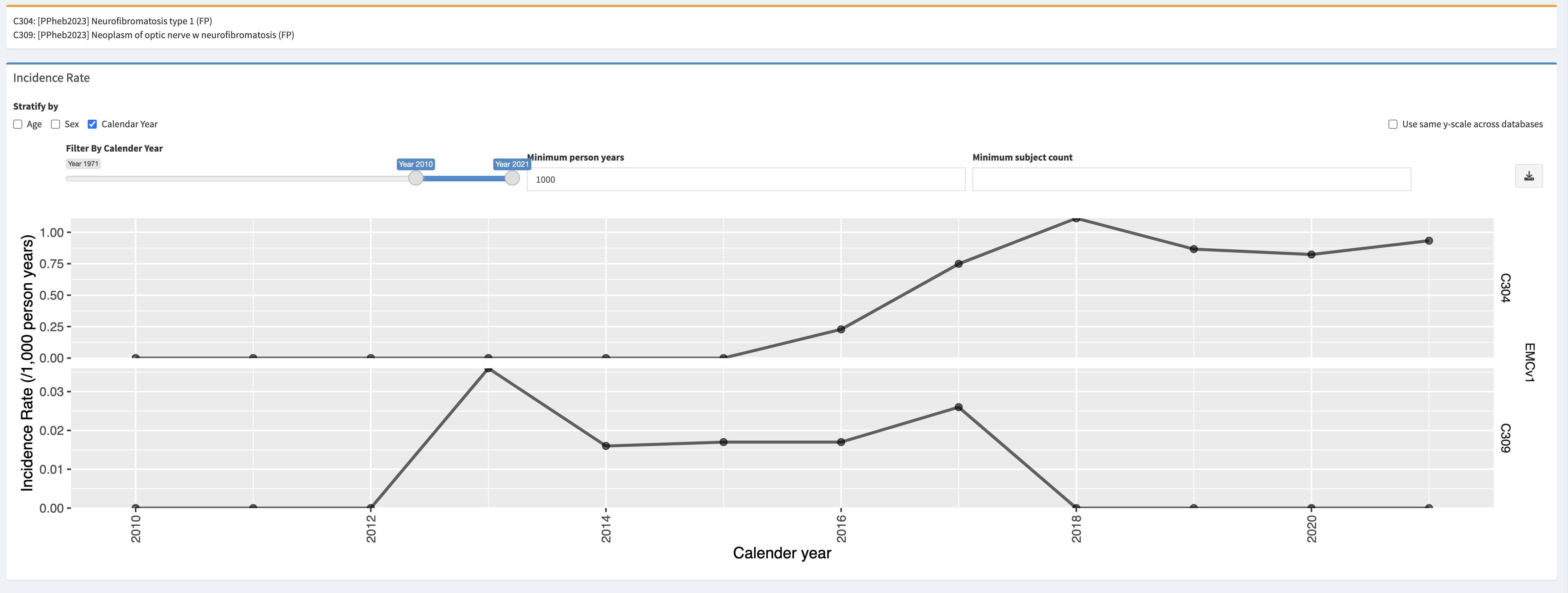
OPGs most frequently develop in young children with NF1 under 7 years of age. OPGs arising in adolescence or adulthood is very rare. Visits to the ophthalmologist are recommended annually for children with NF1 up to 8 years of age. Patients can present with visual acuity loss, unilateral proptosis, visual field defects, strabismus, and optic disc edema (papilledema) or atrophy.
Assessment: NF1 can be diagnosed with the following diagnostic criteria:
A: The diagnostic criteria for NF1 are met in an individual who does not have a parent diagnosed with NF1 if two or more of the following are present:
- Six or more café-au-lait macules over 5 mm in greatest diameter in prepubertal individuals and over 15 mm in greatest diameter in postpubertal individuals (coffee with milk-coloured sports on the skin)
- Freckling in the axillary or inguinal region
- Two or more neurofibromas of any type or one plexiform neurofibroma
- Optic pathway glioma
- Two or more iris Lisch nodules identified by slit lamp examination or two or more choroidal abnormalities (CAs)—defined as bright, patchy nodules imaged by optical coherence tomography (OCT)/near-infrared reflectance (NIR) imaging
- A distinctive osseous lesion such as sphenoid dysplasia, anterolateral bowing of the tibia, or pseudarthrosis of a long bone
- A heterozygous pathogenic NF1 variant with a variant allele fraction of 50% in apparently normal tissue such as white blood cells
B: A child of a parent who meets the diagnostic criteria specified in A merits a diagnosis of NF1 if one or more of the criteria in A are present.
As such, in some cases the diagnosis can be affirmed by physical examination only. In addition, clinicians can choose to do genetic testing, to confirm the diagnosis by detecting a pathogenic mutation in the NF1 gene. The performance of genetic testing has become more prevalent during recent years, the degree to which it is performed will differ between countries and sites. A visit to an ophthalmologist can confirm the presence of iris Lisch nodules or choroidal abnormalities. In addition, an MRI scan can be performed to detect plexiform neurofibroma and brain gliomas, if there is a clinical indication to do so.
The presence of an OPG is confirmed by an MRI scan.
Plan and prognosis: NF1 is a chronic disease, and no treatment exists. Treatment is symptomatic: patients will be treated for manifestations that require it (surgery, chemotherapy, medication). Symptoms of NF1 can be mild, and individuals can live normal and productive lives. However, given the potential for tumour development, long-term surveillance is of importance both in children and in adults. The lifetime risk of cancer in patients with NF1 is estimated to be 60%, compared to 30% in the general population. Children up to 10 generally receive an annual check-up by a paediatrician, neurologist and ophthalmologist. All the children up to 18 years old are generally seen once every two years, and visual screening is recommended. For adults, the schedule of check-ups is less well defined. Screening for breast cancer is recommended yearly for women aged 30-50 years, and per the national guidelines for women older than 50 years.
Once an OPG has been identified, the frequency of neuroimaging and visual assessment depends on the site of the tumour, degree of visual impairment and associated symptoms as well as evidence of progressive disease. The specific intervals of neuroimaging and visual assessments are not well defined, but most centres perform eye examinations and vision testing every three months for the first year after diagnosis, with increasing intervals thereafter. MRI examinations may be performed at similar or less frequent intervals. Approximately 30% of the children with an OPG requires treatment with chemotherapy.
Around 30-50% of the patients with NF1-related OPG will become symptomatic, and ~30% of the patients require treatment. Asymptomatic disease tends to show little progression. While chemotherapy can be effective in shrinking the tumour, it is not always effective in restoring visual acuity.
Literature review
There are a few studies using NF or OPG codes. Some studies used non-specific codes for NF. To note, the coding system in England (ICD10: Q 85.0 Neurofibromatosis) does not distinguish between NF1 and NF2. Any record of schwannoma, meningioma, acoustic neuroma and sensorineural deafness were excluded as researchers assumed these patients had NF2. A Finnish study used these codes (ICD8: 74340, ICD9: 2377A or 2251A) further screened patients for NF1.
One study in US inpatients searched for diagnosis codes for NF1 (237.7, 237.71), neurofibromatosis type 2 (NF2; 237.72), and unspecified neurofibromatosis (NF NOS; 237.70). As NF1 is much more common than NF2, the researchers assumed that NF NOS would nearly always be classified as NF1. These assumptions were tested by comparing the frequency of hospitalizations involving surgery for VS resection in the various groups. Approximately 1 in 6 discharges coded as 237.72 were associated with a VS resection, compared with 0.1% in discharges coded as NF1 or NF NOS, or in the non-NF population. Thus, NF1 codes included NF NOS.
Studies of OPG using the SEER cancer registry in the US generally searched for C72.3 to identify patients with optic nerve as the original tumor site, combined with histologic codes for glioma, either in broad terms (9380-9489, 9380-9442) or more specific 9380 (Glioma), 9382 (Mixed Glioma), or 9400–9421 (Astrocytoma). 50-60% of OPG cases are diagnosed with NF1.
Only one validation study was performed in Canada. The researchers found billing code algorithms had poor performance, likely due to the lack of NF-specific codes. Only 31% of 71 confirmed NF1 cases had hospitalization associated with an ICD code for NF1. A free-text algorithm had high sensitivity 85% (95% CI 74–92%), specificity 100% (95% CI 100–100%), PPV 80% (95% CI 69–88%), NPV 100% (95% CI 100–100%), and a false positive rate of 20% (95% CI 11–33%). Of the 15 false positives, 8 (53%) were possible NF1 cases.
To summarise, based on previous findings, we will aim to test cohort definitions which include NF codes and exclude conditions characteristic of NF2. Given that NF1 is a high risk factor in OPG and NF1 diagnosis codes may not be recorded, especially in specialist settings, we will test a combination of OPG codes +/- NF1 codes.
Cohort Definitions
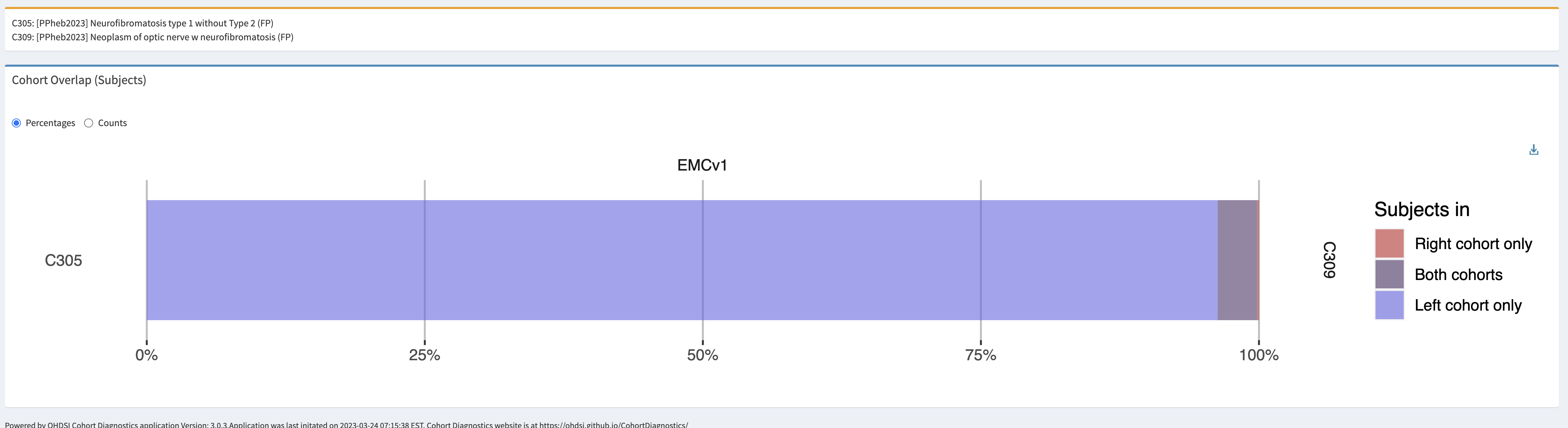
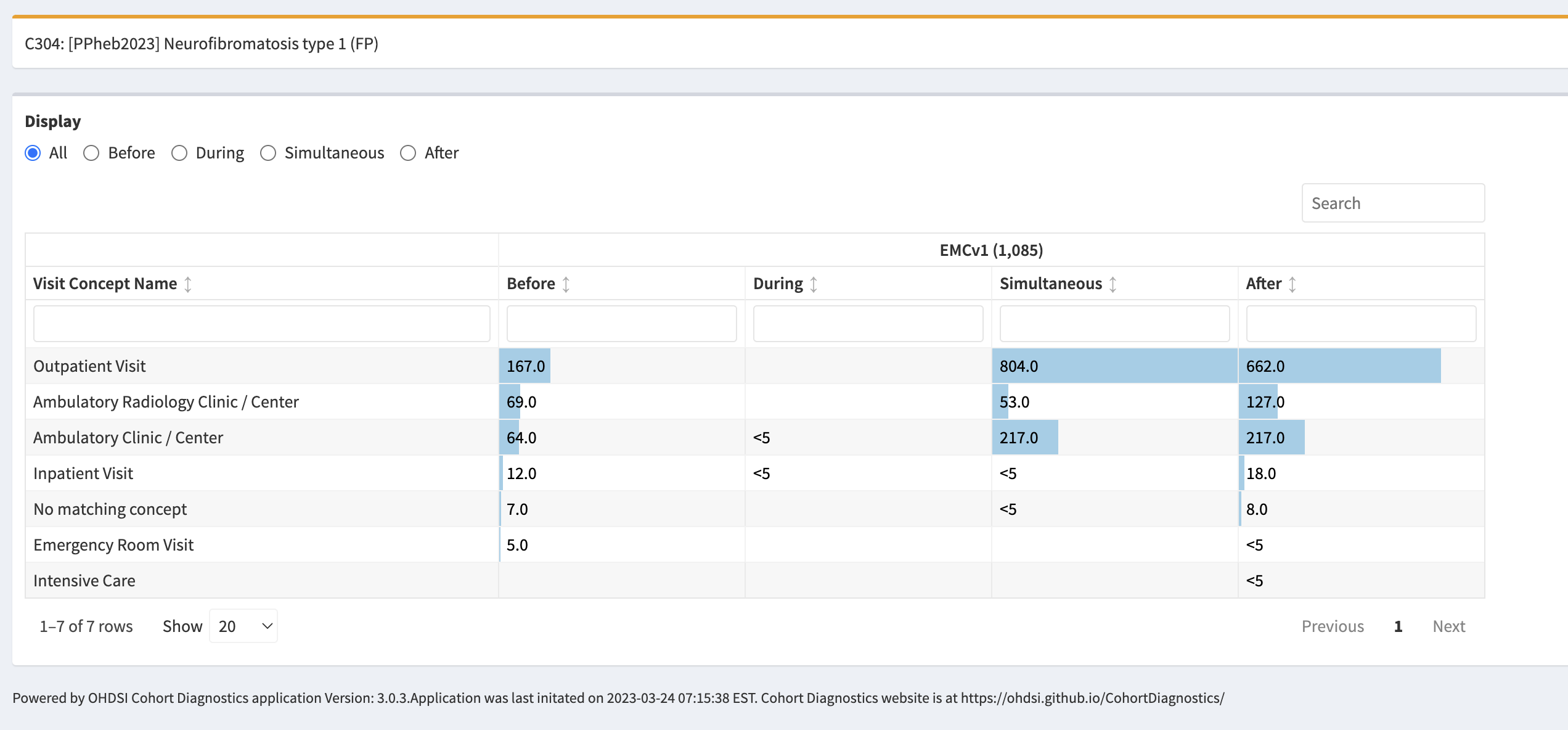
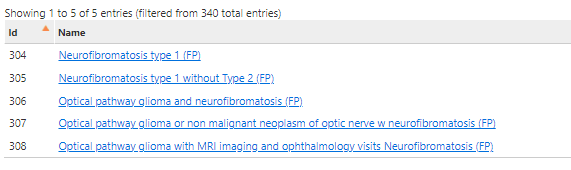
We have defined five cohort definitions. The first two only capture a phenotype for NF1, the last three are specific for the OPG manifestation.
-
Neurofibromatosis type 1
- Logic description: Persons with diagnosis of NF1 diagnosis code (SNOMED:92824003) or descendants (genetic mutation, freckling, elephantiasis, spinal NF, café-au-lait macules, neurofibromas, Noonan syndrome, segmental NF1). These codes are the preferred granular coding for NF1.
-
Neurofibromatosis type 1 – sensitive
- Logic description: Persons with Neurofibromatosis without a diagnosis of NF2, vestibular schwannoma or hearing problems. Not all data sources capture NF subtype (e.g. when using ICD10), so to select NF1 we exclude diagnoses that give a suspicion of NF2 (affecting auditory system).
-
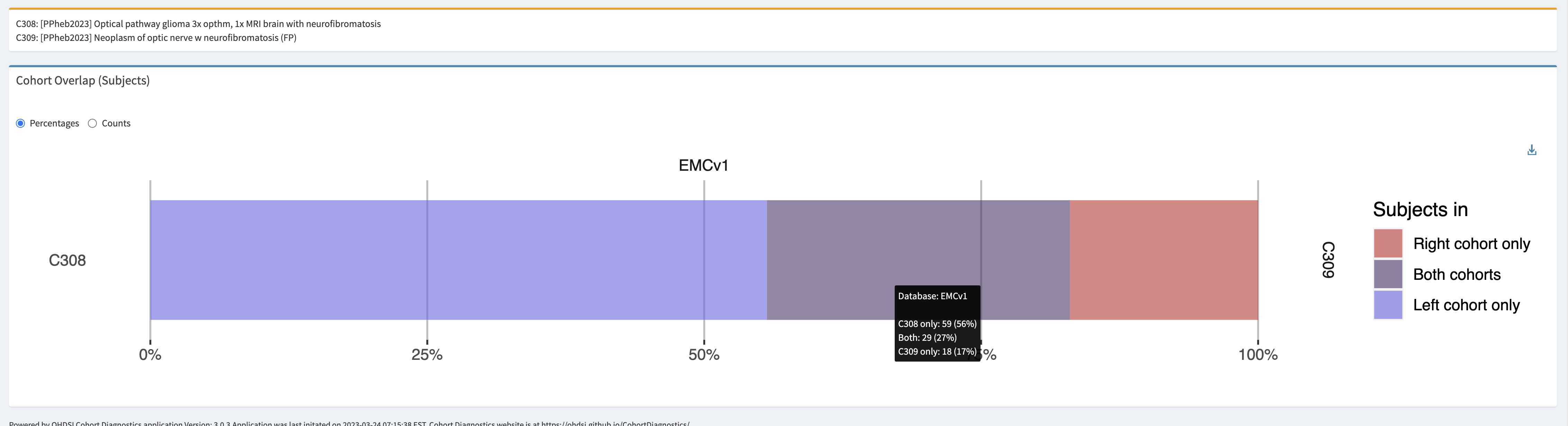
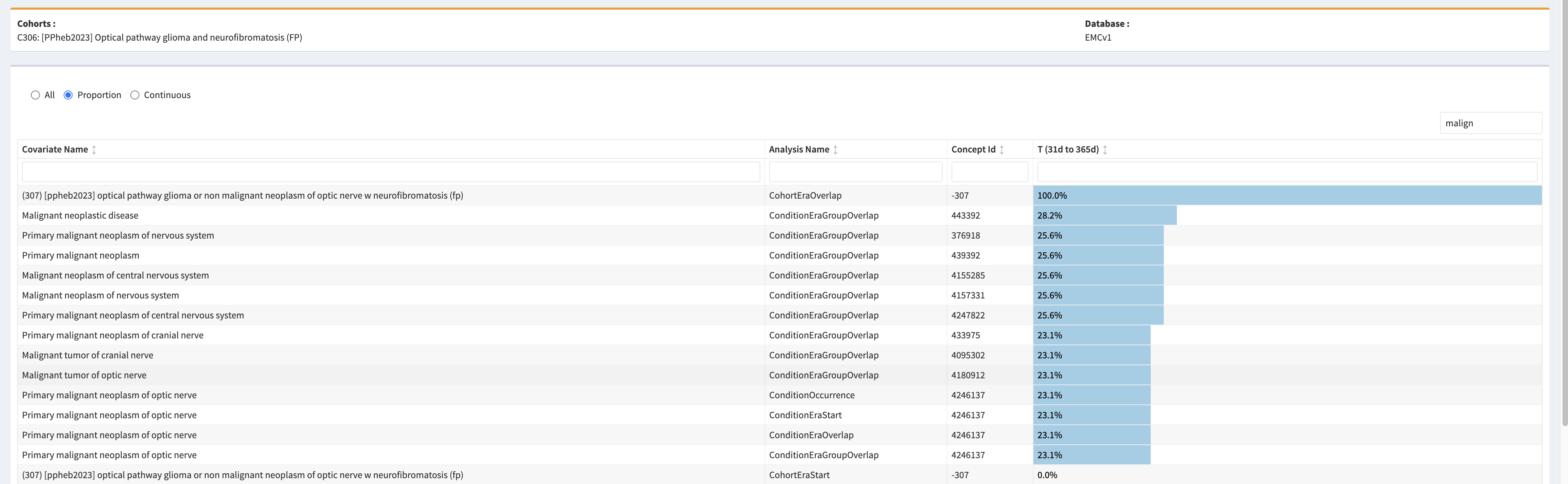
Optical pathway glioma – specific
- Logic description: Persons with an optic nerve glioma and diagnosis of neurofibromatosis anytime in persons history. This would be the preferred code for an OPG.
-
Optical pathway glioma – sensitive
- Updated logic after evaluation, also including malignant neoplasms: Persons with a neoplasm of optic nerve, and a diagnosis of neurofibromatosis anytime in persons history.
-
Optical pathway glioma – follow-up
- Logic description: Persons with neurofibromatosis, that also had one MRI of the brain AND at least 3 ophthalmology visits within one year (anytime in persons history). In some centres, an OPG might be coded as general NF and needs to be inferred from follow-up care after diagnosis.