The t(1;3) is not in any of the Athena vocabularies. I have many other examples and it is expanding. Right now we are only capturing if the test was done. So I guess I just map it to Yes and No?
If we start capturing results how might I go about that? I see some biomarker/ genomics work seems to be happening but slowly.
I ask for making official OHDSI github for further discussion, summarization of requirements, and formal materialization of the genomic extension model. Could you do this, @mvanzandt, @Dymshyts or @Christian_Reich ?
Although not all translocation mutations have 'concept_id’s,
currently you can store the translocation with Yes or No using measurement table like other SNPs.
The contents below are about the various methods and discussion to describe the translocation.
Translocation as “t(A;B)(p1;q2)”
The International System for Human Cytogenetic Nomenclature (ISCN) is used to denote a translocation between chromosomes. The designation t(A;B)(p1;q2) is used to denote a translocation between chromosome A and chromosome B. The information in the second set of parentheses, when given, gives the precise location within the chromosome for chromosomes A and B respectively—with p indicating the short arm of the chromosome, q indicating the long arm, and the numbers after p or q refers to regions, bands and subbands seen when staining the chromosome with a staining dye. The translocation is the mechanism that can cause a gene to move from one linkage group to another.
(Reference: Chromosomal translocation - Wikipedia)
HGVS Nomenclature
The basics of the current HGVS recommendations are explained in a slide presentation from Johan den Dunnen (last updated Apr.2017).
(Reference: http://www.hgvs.org/varnomen/HGVS-basics2017.pdf)
Translocation in HGVS Nomenclature
There was a discussion in HGVS2013 about the method to describe translocations.
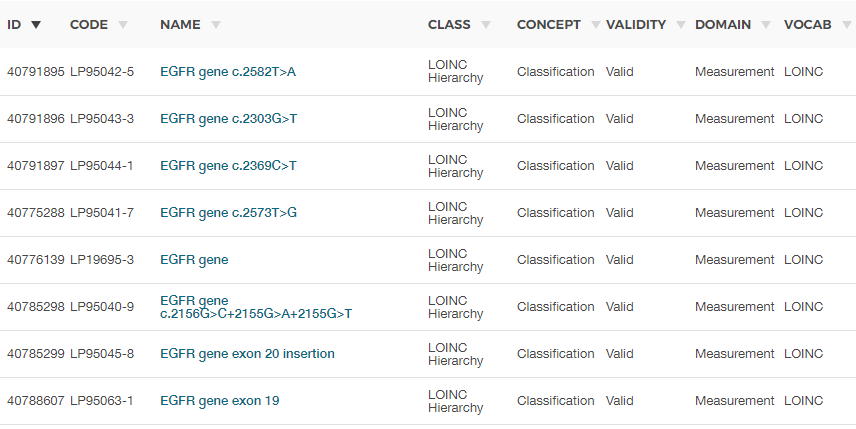
So I have been using yes/no for if the test shows the translocation. Then there are translocations such as t(11;16) that aren’t in any of the current Athena dictionaries. What approach do I take for these instances. In the Immuno-oncology space the number of biomarkers/ genetic tests is expanding faster than things like LOINC can keep up with.
In the era of Next Generation Sequencing (NGS) rather than Sanger Sequencing, ‘variant’ is hard to be described with ‘concept_id’. Because the lesser-known variants are much more than the famous variants.
Basically, the ‘variant’ is a myriad of combinations of 1) POSITIONs in DNA where sequence changes occur and 2) NUCLEOTIDEs(A,T,G,C) before and after modification.
It would be better to separate and record ‘gene’ and ‘change’ information than to define the infinite number of ‘gene + change’ cases as concept_id.
About 40,000 human gene lists can be standardized through the HUGO Gene Nomenclature Committee (HGNC).
The ‘change’ information is to create a rule that records in string data type according to the Human Genome Variation Society (HGVS) nomenclature.
At present, many translocation variants will not have concept_id in Athena.
All you can do is request a new concept_id or temporarily assign your own concept_id.
Alternatively, your data can be stored using the Genomic-CDM if you need although it’s not an official way yet.
Let me read all this and attempt to get my head around it. I need to see how to implement the Genomic CDM and Oncology internally in our systems. We are moving to a semantic analytic tool that we could utilize this in potentially.
Okay, So my genetics training is weak at best. But let me try to use some examples:
So currently LOINC has a code for:
t(6;9).
We only ask if a test for t(6;9) is done and the results (pos/neg)
Concept Name: t(6;9)(p22;q34)(DEK,NUP214) fusion transcript [Presence] in Blood or Tissue by Molecular genetics method
This should go into the CDM definition in the new repository. The problem is this: There are some well-known chromosomal aberations, and LOINC has a good list. And there is an infinite amount of possible aberrations, for which such a notation would be useful. And there is a gray zone in the middle, aberrations that appear non-nonsense and are under research. The CDM needs to deal with these.
Because the involved genes may be irrelevant or not defined. Often, these translocation create a specific aberation that fuses two independent genes, or combines a transcription factor binding site (promotor or repressor region) with a gene for which it was not designed. But again, in many cases these translocations are not precisely defined, and become disease attributes in their own right.
Does that help?
We do have to figure that out in the above mention Genomic WG. There is no shortcut here, @jliddil1.