
I’d be interested in joining
Not sure if this forum is still accepting new members. I have just joined OHDSI, and would like to be part of the CDM Genomics WG.
All Working Groups, like any community activities in OHDSI are always open as a matter of principle. So, welcome to the family. You are in. 
Thank to help of @clairblacketer, we’ve made wiki page for genomic CDM WG.
In the wiki page, the pilot model for genomic CDM was posted and some issues or comments were also posted.
We’ve converted genomic data of NGS to this model from one hospital(Ajou university). We are trying to converted TCGA open dataset into this mode, too.
I develop a machine learning algorithm which combining clinical features from existing CDM and genomic features from G-CDM by using FeatureExtraction and PatientLevelPrediction package. The code will be soon released, too.
We’re now working on summarizing what we’ve discussed in this thread. Until now, we’ve just suggested the prototype for the great challenge to unify genomic data around the world.
We need your thorough reviews. I do appreciate everyone for their passionate participation and comments.
I’d also be interested in participating here. Thanks.
1 Like
I’d be interested in joining too!
Is the Genomic CDM Subgroup still moving forward? We have interest at Northwestern in implementing the Genomic proposal extension.
It takes time for us to develop standardized ontology system for functional and structural meanings of genetic variants.
We’re now trying to validate G-CDM by comparing Ajou university’s lung cancer mutation data and TCGA’s lung cancer mutation data. We’re building a kind of genetic analytic pipeline, based on OHDSI ecosystem so that researchers can easily analyze the genetic profiles between cohorts produced by ATLAS.
I apologize for my laziness. We’ll propose new version (not much changed from former version. We’ve focused on ontology system for representing variants) and the demo of our experiments within one or two weeks. I want to start G-CDM WG meeting again after that.
@mgurley , I am so interested to hear that! Please let me know if you need anything about this!
An updated version of G-CDM was released in Google Drive.
There are Specification and Sample Data of G-CDM. You can also see TCGA (The Cancer Genome Atlas) Sample Data and how it can be transform to G-CDM (ETL specification).
In this version, we applied standardized ontology system for representing structural and functional meanings of genetic variants. Because of a discrepancy in vocabulary for variant between databases, it needed to mapping each data source value to concept name and id.

Using hierarchical ontology system of “Sequence Ontology”, different expression of variant type in each database unified to common vocabulary.
I would like to present and discuss with you in Genomic-CDM Working Group and Teleconference.
4 Likes
Hi all,
thanks a lot for your effort to extend OMOP to adopt different data sources. Is this working group still active? It would be really interesting to evaluate further extensions to the OMOP CDM to accommodate genomics datasets.
Sure!
We are working on developing the genomic-CDM (G-CDM) as an extension model of OMOP-CDM.
There was an Oncology+Genomic working group face to face meeting in in 2018 OHDSI Symposium (Bethesda).
Here is our Google drive link. You can see a specification and presentation materials about G-CDM which is on developing.
We are on tunning the schedule for the online meeting for the next steps.
Thank you for your interest!
1 Like
Just went through Drive documents. I admit it is an ambitious goal to put genomic data to structured database model not only because of the data size, but also because of the non-standardized notation used for genomic data. However, there is still a strong need for a good database format for that.
We have just calculated pharmacogenomics recommendations for 44,000 biobank participants (https://www.nature.com/articles/s41436-018-0337-5) and I only wanted to highlight that information about genotype PHASING (are the genotypes of 2 SNPs phased) and IMPUTATION (was the genotype imputed, what was the value probability) are also very important pieces of information when you are dealing with genomic data.
1 Like
Thank you for your information about “Phasing” and “Imputation” for dealing sequencing data.
I’ll review that issue and let G-CDM adopt those concepts in adequate position and method!
Thanks again 
Hello. Is [CDM Builders] the best place to follow the oncology/genomics CDM extension discussions, or are there different discussion groups for those topics? Thank you.
@jmethot: Go ahead. There is not a ton of strict rules here. CDM Builders is just fine.
What’s your question?
I would like to join as well.
Agenda: OMOP-CDM Extension for NGS Data
First of all, I appreciate for all your interest and discussion to support the genomic CDM Workgroup.
These days we are thinking basic concepts; why we need (do we need) a table extension for genomic data; which elements of genomic data has to be stored in OMOP-CDM; how the genomic CDM can be used in clinical practice.
Why is genetic material important in the clinical practice?
- The patient’s genomic data are used as an indicator for determining the cancer stage and anticancer treatment according to the NCCN guidelines (Figure 1).


Figure 1. Genomic alteration data used in clinical decision
The way to store the genomic data in current OMOP-CDM
-
Currently, OMOP-CDM is a structure that stores only a few well-known mutations.
In the Measurement/Observation table, the combination of [gene name + sequential variant + specimen + inspection method + exon number + variant type] information is created in one set and has a concept ID (Figure 2).
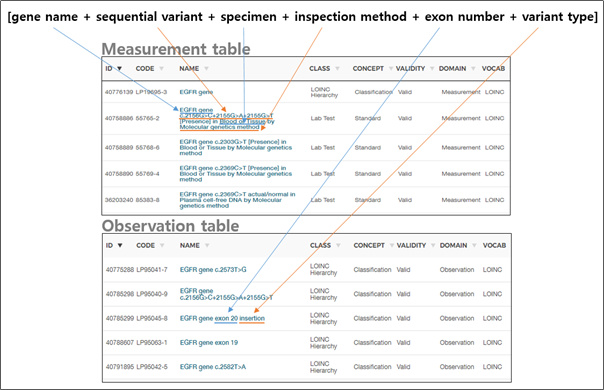
Figure 2. How to present a variant in the current OMOP-CDM -
This is an efficient way to store the result of only a few typical mutations, such as a single gene or dozens/hundreds of specific mutations, to express whether or not those mutations are present.
Why does OMOP-CDM need to be expanded?
Reason 1: The number of variant subject to sequencing has increased exponentially.
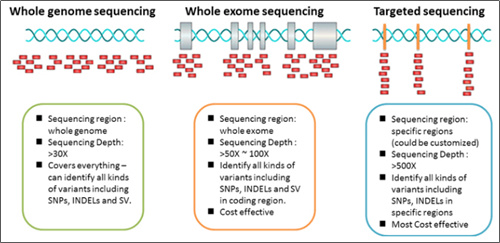
Figure 3. Types of next generation sequencing (NGS)
*Image source: https://2wordspm.wordpress.com/2017/10/30/ngs-검사-whole-genome-exome-targeted-sequencing-비교/
-
For the recently generalized Targeted Next Generation Sequencing (Targeted-NGS) technology, the number of genes to be examined is among tens and hundreds, and each gene has its own area for testing.
-
However, countless variations can occur at a sequence (A), such as deletions (A to -), substitution (A to T / C / G), and insertions (A to AT / ATG / ATGC / ATGCC / ATGCCTTACGGAT and so on……).
-
It is impossible to make the number of all these cases into one set, as it is now.
-
In particular, Whole Exome Sequencing (WES) and Whole Genome Sequencing (WGS) technologies examine entire exome and genome sequences, respectively, and are being used to find patients who can be treated with certain drugs as it has been found that Tumor Mutation Burden (TMB)* tests in immune diseases are related to predicting Immune checkpoint inhibitor (ICI) treatment effects (Figure 4). (DOI: 10.1200/JCO.2017.75.3384 Journal of Clinical Oncology 36, no. 7 (March 1 2018) 633-641.)
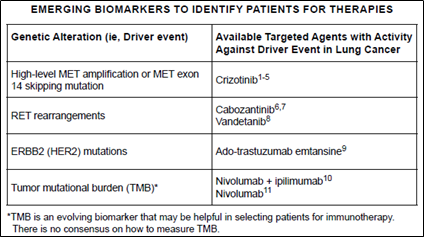
Figure 4. TMB as an emerging biomarker in NCCN Guidelines Version 1.2019 Non-Small Cell Lung Cancer
*TMB (Tumor Mutation Burden): TMB is the calculation of the number of mutations in cancer cells.
The higher the TMB, the higher the response to immunosuppression drugs is shown.
- Because WES and WGS technologies examine the entire exome and genome sequence, it is inefficient to create all the variation concepts as they are today, as with targeted-NGS.|
Reason 2. Non-variant information is also required to interpret NGS testing.
-
In addition to the current measurement/observation table, there are features that should be taken into account when comparing multi-center data of NGS testing.
-
This is because the method of the NGS testing is not standardized like other clinical lab test tests.
-
They should be recorded/standardized together to interpret NGS testing between institutions to further classify the patient’s genetic variation and cancer condition and to identify the outcome of clinical care.
-
Currently, most of these data may be retrieved from pathology reports or from EMR, even though it’s very difficult, but in CDM, there is no way to obtain the well refined NGS results.
| > Non-vairant features to be recorded/standardized |
-
Sequencing platform (device & software) information
: Information about the name and version of the platform using sequencing assigned by the institution. -
Reference genome
: Prior knowledge that is used in aligning the leads to recognize changes in sequence. Because the reference genome acts as a comparison criterion, it is important to confirm whether the reference genome is identical or not when comparing variants between institutions. -
Read depth (variant & total)
: Reads are the thousands of pieces of nucleic acid that you analyze when you run an NGS. NGS amplifies the reads and sequencing them over and over again, supplementing slightly incorrect parts to increase the accuracy. Read depth is the number of times a single location has been read during sequencing. Bigger read depth means better accuracy. You should exclude variants with read depth under threshold during analysis for increasing accuracy of comparison. -
Genotype
: Whether the variant comes from somatic or germline -
Annotation information
: The clinical impact of the variant
Things you can do with Genomic-CDM (Use cases)
-
Identifying patients for therapies
: By using variants data resulted from NGS, you can figure out how many patients are adequate for immunotherapy (ex. Nivolumab) in each centers by calculating tumor mutational burden (TMB) from the result data of whole exome sequencing (WES). -
Selecting patients for clinical trials
: By using variants data and linked clinical data, you can find and encourage patients to involve certain clinical trials (ex. NCT02296125) who have specific mutation profiles and treatment history. -
Finding genes/variants related to outcomes
: By using variants characteristic and linked drug/treatment exposure data, you can discover genomic characteristic of patient group having a poor reaction to drugs.
- You can also use non-variant information of the genomic-CDM in…
- confirming the comparability of the genomic data between institutions by using information of sequencing platform, reference genome, genotype.
- filtering the genomic data by quality by using information of read depth of variant and total target read.
The meeting of the genomic CDM working group has been suspended some while, but we will hold the meeting again soon. We look forward to your participation. 
5 Likes
Interested
Friends:
That is very good. Thanks for putting new life into it.
Because we do need to get to the bottom of this problem. So far, we have mostly collected a pile of potential pieces of information, which could be useful. The problem with that approach is that we may spin and spin with little results:
- We have no standard model
- We have not enabled network research
- We have not enabled or developed standardized, systematic or scaling methods
As a result, we are still far away from generating scientific insights.
The only chance I see is to work this from the top down:
Start with the use cases
This is a good starting point. But we need to agree on them. Even if we have a longer list with high and low priority it would help tremendously.
Personally, I would split the last one into
- Utilize established genes/variants in estimation/prediction/diagnostic models
- Establish (explore and identify) genes/variants for estimation/prediction/diagnostic purposes
We also need to declare what we are not going to tackle. For example, building a genomic pipeline from raw data.
Once we have these, we continue with
Building the model
- What data about patients do we need for these use cases, and how do we represent them. There is good work available from @ShinSeojeong, @1118 (we should not have two concepts for the same person, btw, violation of OMOP CDM
 ), @KKP1122 and @Yurang_Park. But we need to come up with a compact and efficient solution. Not all data, just because they exist, should be standardized. Only if they serve the use cases.
), @KKP1122 and @Yurang_Park. But we need to come up with a compact and efficient solution. Not all data, just because they exist, should be standardized. Only if they serve the use cases. - We need to outsource all prior knowledge, like known variants and their relevance, into the vocabularies, and take it out of the data
- We need to standardize the representation of the data in the vocabularies
This should be an iterative process. V1 should the minimal model to make progress against the use cases. Once we have that, we need to:
Put some data into V1
Test them against use cases
Expose the results to the community
Before that, we should not go into V2.
Another rule we have successfully applied to OMOP CDM and Vocabulary development is the notion of “theft”. We should not invent anything that exists and is established. We should evaluate existing systems and adopt, instead of creating new ones. That will require some work. I can help with vocabularies. @1118’s mention of the NCCN is a first good step.
Makes sense?
That remark made my day, @SCYou.
1 Like
So since we are dong WES, RNA, WGS how can I map these now in OMOP CDM?